L’hypertension artérielle pulmonaire (HTAP) est une maladie rare caractérisée par un remodelage intense des artères pulmonaires musculaires de petit calibre aboutissant à l’élévation des résistances vasculaires pulmonaires (RVP) et de la pression artérielle pulmonaire (PAP), avec pour conséquences l’apparition d’une insuffisance cardiaque droite et le décès.
La prise en charge de l’HTAP s’est considérablement améliorée au cours des dernières années parallèlement aux progrès réalisés dans la compréhension des mécanismes de la maladie, au développement de médicaments innovants et à l’application de stratégies thérapeutiques plus agressives. Cette prise en charge associe des mesures générales, des traitements symptomatiques et des thérapeutiques dites « spécifiques » ciblant l’équilibre vasoconstriction/vasodilatation et proprolifération/antiprolifération des artères pulmonaires. L’HTAP étant une maladie rare, sa prise en charge doit être assurée dans un centre d’expertise en hypertension pulmonaire (HTP).1 En France, elle est assurée au sein d’un réseau labélisé de centres de référence et de compétence maladies rares (CRMR PulmoTension).
La prise en charge de l’HTAP s’est considérablement améliorée au cours des dernières années parallèlement aux progrès réalisés dans la compréhension des mécanismes de la maladie, au développement de médicaments innovants et à l’application de stratégies thérapeutiques plus agressives. Cette prise en charge associe des mesures générales, des traitements symptomatiques et des thérapeutiques dites « spécifiques » ciblant l’équilibre vasoconstriction/vasodilatation et proprolifération/antiprolifération des artères pulmonaires. L’HTAP étant une maladie rare, sa prise en charge doit être assurée dans un centre d’expertise en hypertension pulmonaire (HTP).1 En France, elle est assurée au sein d’un réseau labélisé de centres de référence et de compétence maladies rares (CRMR PulmoTension).
Mesures générales et traitements symptomatiques
Les mesures générales et les traitements symptomatiques de l’HTAP reposent sur la prévention des situations à risque et la prescription de traitements visant à réduire les symptômes.
Activité physique et réentraînement
Des études récentes ont montré un impact du réentraînement à l’exercice sur les capacités à l’effort et la qualité de vie dans l’HTAP.2 Un réentraînement à l’effort supervisé est donc recommandé pour les patients dont la maladie est bien contrôlée sous traitement de l’HTAP (recommandation de classe IA).1
Grossesse et contraception
La grossesse aboutit à des modifications hémodynamiques, essentiellement au cours du troisième trimestre, qui peuvent entraîner une décompensation cardiaque droite possiblement fatale pour la mère et l’enfant. Dans ce contexte, la grossesse est fortement déconseillée chez les femmes atteintes d’HTAP.1 Selon les dernières recommandations, les femmes en âge de procréer et leurs partenaires doivent être informés par un médecin spécialisé en HTP des risques et de l’issue incertaine en cas de grossesse. Une contraception efficace est préconisée, d’autant plus que certains traitements spécifiques comme les antagonistes des récepteurs de l’endothéline (ARE) ont un effet tératogène.
Anesthésie et chirurgie
Toute anesthésie ou intervention chirurgicale est susceptible d’aboutir à des complications possiblement graves. Il faut donc discuter du rapport bénéfice-risque des interventions programmées et privilégier, lorsque cela est possible, des interventions sous anesthésie locorégionale. Quoi qu’il en soit, en cas d’anesthésie envisagée, une prise en charge multidisciplinaire dans un centre spécialisé en HTP est recommandée.1
Prévention des infections
Les infections peuvent être à l’origine d’une aggravation clinique de l’HTAP. Les vaccinations antigrippale, antipneumococcique et anti-SARS-CoV-2 sont ainsi recommandées pour tous les patients.1
Altitude et hypoxie liée aux transports aériens
Les séjours en altitude au-delà de 1 500 mètres sont déconseillés ainsi que les transports aériens en cabine non pressurisée.1 En effet, l’hypoxémie survenant en altitude est la conséquence d’une baisse de la pression atmosphérique et ainsi de la pression inspiratoire en oxygène. Elle peut, notamment lorsqu’elle est prolongée, favoriser une aggravation de l’HTP par un phénomène de vasoconstriction pulmonaire hypoxique.
Anticoagulation au long cours
L’anticoagulation orale au long cours, autrefois considérée comme indispensable au cours de l’HTAP, ne doit plus être proposée qu’au cas par cas chez les patients présentant une HTAP idiopathique, familiale ou associée à une prise d’anorexigènes (recommandation de classe IIb-C).1 En effet, celle-ci n’a pas montré de bénéfice en matière de survie dans l’HTAP idiopathique.3 Elle est même délétère dans d’autres formes d’HTAP, comme la sclérodermie systémique (associée à une surmortalité). Les anticoagulants doivent donc être envisagés uniquement en cas d’indication autre que l’HTAP elle-même (fibrillation atriale, maladie thromboembolique…).
Traitement diurétique
La défaillance cardiaque droite aboutit à une rétention hydrique, avec augmentation de la pression veineuse centrale, congestion hépatique et œdèmes périphériques. La pratique clinique montre que le traitement diurétique (diurétiques de l’anse ou antialdostérone) permet souvent d’améliorer les symptômes (recommandation classe IC).1 L’adaptation de la posologie des diurétiques est guidée par les signes cliniques (poids, présence de signes d’insuffisance cardiaque droite comme les œdèmes des membres inférieurs) et l’évaluation hémodynamique ou échocardiographique (morphologie de la veine cave inférieure) des pressions de remplissage droites.
Oxygénothérapie
La prescription de l’oxygénothérapie est souvent calquée sur ce qui est pratiqué dans l’insuffisance respiratoire chronique associée à une bronchopneumopathie chronique obstructive (BPCO) : celle-ci est prescrite lorsque la pression partielle en oxygène (PaO2) est inférieure à 60 mmHg au repos (recommandation de classe IC).1 Elle doit être prescrite au moins 16 à 18 heures par jour mais elle peut être prise 24 heures sur 24. Il n’y a pas de recommandation pour la prescription d’oxygénothérapie à l’effort. Enfin, le bénéfice de l’oxygénothérapie sur la survie n’est pas démontré dans l’HTAP.
Inhibiteurs calciques
Chez certains patients, la vasoconstriction constitue le mécanisme essentiel de l’HTAP. Ces patients sont identifiés par un test de vasoréactivité en aigu réalisé lors du cathétérisme cardiaque droit initial, le plus souvent par inhalation de monoxyde d’azote (NO), mais il est également possible de le réaliser à l’iloprost inhalé ou à l’époprosténol par voie intraveineuse. Les patients « répondeurs » au test de vasoréactivité sont éligibles à un traitement par inhibiteurs calciques en monothérapie.4 La réponse clinique et le pronostic de ces patients sont habituellement excellents. Néanmoins, la proportion de patients répondeurs en aigu est très faible, représentant environ 10 % des patients avec une HTAP idiopathique, héritable ou associée à la prise d’anorexigènes ; aucune réponse n’ayant été observée dans les autres formes d’HTAP. Dans ce contexte, le test de vasoréactivité en aigu n’est recommandé que dans ces trois formes d’HTAP.
De plus, même si une majorité de patients répondeurs en aigu gardent une amélioration clinique et hémodynamique au long cours (définie par le maintien en classe fonctionnelle [CF] I ou II de la New York Heart Association [NYHA], associée à une amélioration hémodynamique prolongée après au moins un an de traitement sous inhibiteurs calciques seuls), permettant de poursuivre un traitement par inhibiteurs calciques seuls, certains patients échappent à ce traitement simple (et donc nécessitent un renforcement thérapeutique) sans qu’on en connaisse vraiment les facteurs de risque. La posologie des inhibiteurs calciques utilisés dans cette indication (amlodipine, félodipine, nifédipine ou diltiazem) est généralement supérieure à celle habituellement prescrite dans d’autres indications comme l’hypertension artérielle systémique.1
De plus, même si une majorité de patients répondeurs en aigu gardent une amélioration clinique et hémodynamique au long cours (définie par le maintien en classe fonctionnelle [CF] I ou II de la New York Heart Association [NYHA], associée à une amélioration hémodynamique prolongée après au moins un an de traitement sous inhibiteurs calciques seuls), permettant de poursuivre un traitement par inhibiteurs calciques seuls, certains patients échappent à ce traitement simple (et donc nécessitent un renforcement thérapeutique) sans qu’on en connaisse vraiment les facteurs de risque. La posologie des inhibiteurs calciques utilisés dans cette indication (amlodipine, félodipine, nifédipine ou diltiazem) est généralement supérieure à celle habituellement prescrite dans d’autres indications comme l’hypertension artérielle systémique.1
Lire aussi | Consensus sur l’hypertension pulmonaire
Traitements « spécifiques » de l’HTAP
Les thérapies actuelles de l’HTAP ont pour cible la dysfonction endothéliale des artères pulmonaires. Celle-ci se traduit par un défaut de production de substances vasodilatatrices et antiproliférantes (prostacycline [PGI2] et NO) et par un excès de production de substances vasoconstrictrices et proproliférantes (endothéline-1 [ET-1]). Ces modifications conduisent à une vasoconstriction excessive et une prolifération des cellules musculaires lisses des petites artères pulmonaires. La plupart des thérapeutiques ciblées de l’HTAP ont été évaluées dans des études randomisées, contrôlées contre placebo, à court terme (de trois à quatre mois), avec comme critère principal de jugement l’amélioration des capacités à l’effort évaluée par la distance parcourue au test de marche de six minutes (TM6). Les études plus récentes ont évalué le délai avant aggravation clinique à plus long terme selon un critère de jugement composite.
On distingue trois voies thérapeutiques principales.
On distingue trois voies thérapeutiques principales.
Voie de la prostacycline
La PGI2, principal dérivé de l’acide arachidonique, est produite par les cellules endothéliales. Elle entraîne une augmentation de la concentration en adénosine monophosphate cyclique (AMPc) dans les cellules musculaires lisses, à l’origine d’une relaxation, d’une vasodilatation et d’un effet antiproliférant.
Les analogues de la PGI2 disponibles en France sont l’époprosténol et le tréprostinil.
L’époprosténol doit, du fait de sa demi-vie très courte (environ trois minutes), être administré par voie intraveineuse continue à l’aide d’une pompe connectée à un cathéter veineux central en position sous-clavière ou jugulaire, tunnelisé sous la peau. Il a été montré que ce traitement permet une amélioration clinique et hémodynamique mais aussi de la survie des patients.5-7 Les effets indésirables sont fréquents et dose-dépendants, avec notamment des douleurs des mâchoires, des diarrhées, des céphalées ou des nausées. Les complications les plus sévères sont liées à la présence d’un cathéter veineux central à demeure, avec en particulier des infections locorégionales ou systémiques. Ainsi, malgré sa complexité et les risques infectieux, l’époprosténol reste le traitement de référence des formes sévères d’HTAP.1
Le tréprostinil est une alternative, avec une demi-vie plus longue que celle de l’époprosténol. Il est également administré de manière continue mais par voie sous-cutanée à l’aide d’une pompe, similaire aux pompes à insuline. L’intérêt du tréprostinil, par rapport à la voie intraveineuse, est l’absence de risque infectieux et la possibilité de courtes périodes de débranchement du dispositif. Il a été montré que ce traitement améliore les capacités d’exercice, les symptômes et l’hémodynamique des patients en CF II à IV de la NYHA.8 Ses principaux inconvénients sont des douleurs et inflammations locales, au point d’injection, parfois majeures.
Le sélexipag, agoniste oral des récepteurs IP de la PGI2, est approuvé dans l’HTAP depuis 2016, en particulier chez des patients qui restent à risque intermédiaire faible, en association avec un ARE et un inhibiteur de la phosphodiestérase de type 5 (IPDE-5).1 Son efficacité sur le ralentissement de la progression de la maladie a été démontrée.9 Les effets indésirables de ce médicament (similaires à ceux de la prostacycline injectable) sont fréquents, ce qui nécessite une adaptation individuelle de la posologie.
Les analogues de la PGI2 disponibles en France sont l’époprosténol et le tréprostinil.
L’époprosténol doit, du fait de sa demi-vie très courte (environ trois minutes), être administré par voie intraveineuse continue à l’aide d’une pompe connectée à un cathéter veineux central en position sous-clavière ou jugulaire, tunnelisé sous la peau. Il a été montré que ce traitement permet une amélioration clinique et hémodynamique mais aussi de la survie des patients.5-7 Les effets indésirables sont fréquents et dose-dépendants, avec notamment des douleurs des mâchoires, des diarrhées, des céphalées ou des nausées. Les complications les plus sévères sont liées à la présence d’un cathéter veineux central à demeure, avec en particulier des infections locorégionales ou systémiques. Ainsi, malgré sa complexité et les risques infectieux, l’époprosténol reste le traitement de référence des formes sévères d’HTAP.1
Le tréprostinil est une alternative, avec une demi-vie plus longue que celle de l’époprosténol. Il est également administré de manière continue mais par voie sous-cutanée à l’aide d’une pompe, similaire aux pompes à insuline. L’intérêt du tréprostinil, par rapport à la voie intraveineuse, est l’absence de risque infectieux et la possibilité de courtes périodes de débranchement du dispositif. Il a été montré que ce traitement améliore les capacités d’exercice, les symptômes et l’hémodynamique des patients en CF II à IV de la NYHA.8 Ses principaux inconvénients sont des douleurs et inflammations locales, au point d’injection, parfois majeures.
Le sélexipag, agoniste oral des récepteurs IP de la PGI2, est approuvé dans l’HTAP depuis 2016, en particulier chez des patients qui restent à risque intermédiaire faible, en association avec un ARE et un inhibiteur de la phosphodiestérase de type 5 (IPDE-5).1 Son efficacité sur le ralentissement de la progression de la maladie a été démontrée.9 Les effets indésirables de ce médicament (similaires à ceux de la prostacycline injectable) sont fréquents, ce qui nécessite une adaptation individuelle de la posologie.
Voie de l’endothéline-1
L’ET-1 agit par liaison à deux récepteurs, de type A (ET-A) et de type B (ET-B).
Deux antagonistes des récepteurs de l’ET-1 (administrés par voie orale) sont disponibles en France : le bosentan, antagoniste des deux récepteurs ET-A et ET-B, et l’ambrisentan, antagoniste sélectif des ET-A.10,11 Le macitentan, autre antagoniste des ET-A et ET-B, a montré son efficacité sur la réduction des événements de morbi-mortalité au long cours dans une grande étude multicentrique.12 Bien qu’ayant obtenu une autorisation de mise sur le marché à l’échelon européen, il n’est pas remboursé ni disponible en France.
La tolérance clinique des ARE est habituellement bonne. Un des principaux effets indésirables du bosentan est une augmentation des enzymes hépatiques, survenant dans environ 5 à 10 % des cas et réversible à l’arrêt du traitement. La rétention hydrosodée parfois observée avec ces médicaments est un effet indésirable de classe, néanmoins plus fréquemment observé avec l’ambrisentan. Enfin, il faut être attentif aux interactions médicamenteuses avec le bosentan, qui est un inducteur du CYP450.
Deux antagonistes des récepteurs de l’ET-1 (administrés par voie orale) sont disponibles en France : le bosentan, antagoniste des deux récepteurs ET-A et ET-B, et l’ambrisentan, antagoniste sélectif des ET-A.10,11 Le macitentan, autre antagoniste des ET-A et ET-B, a montré son efficacité sur la réduction des événements de morbi-mortalité au long cours dans une grande étude multicentrique.12 Bien qu’ayant obtenu une autorisation de mise sur le marché à l’échelon européen, il n’est pas remboursé ni disponible en France.
La tolérance clinique des ARE est habituellement bonne. Un des principaux effets indésirables du bosentan est une augmentation des enzymes hépatiques, survenant dans environ 5 à 10 % des cas et réversible à l’arrêt du traitement. La rétention hydrosodée parfois observée avec ces médicaments est un effet indésirable de classe, néanmoins plus fréquemment observé avec l’ambrisentan. Enfin, il faut être attentif aux interactions médicamenteuses avec le bosentan, qui est un inducteur du CYP450.
Voie du NO
Les IPDE-5, sildénafil et tadalafil, administrés par voie orale, entraînent une augmentation de la concentration intracellulaire de guanosine monophosphate cyclique (GMPc) ayant pour conséquence une relaxation du muscle lisse vasculaire pulmonaire ainsi qu’une inhibition de la prolifération des cellules musculaires lisses.13,14 Administrés en monothérapie ou en association à d’autres traitements de l’HTAP, ils améliorent les symptômes, les capacités d’exercice et l’hémodynamique des patients ayant une HTAP.13,14
Le riociguat est un stimulateur de la guanylate cyclase soluble, stimulant la synthèse du GMPc indépendamment de la production endogène de monoxyde d’azote (NO). Ce médicament utilisable par voie orale a montré son efficacité à la fois dans l’HTAP et l’HTP thromboembolique chronique.15,16
Le riociguat est un stimulateur de la guanylate cyclase soluble, stimulant la synthèse du GMPc indépendamment de la production endogène de monoxyde d’azote (NO). Ce médicament utilisable par voie orale a montré son efficacité à la fois dans l’HTAP et l’HTP thromboembolique chronique.15,16
Stratégie thérapeutique
Après confirmation du diagnostic d’HTAP dans un centre expert, les mesures générales incluant l’éducation thérapeutique du patient sont mises en place et les traitements symptomatiques (diurétiques, oxygénothérapie…) sont entrepris si nécessaire.1
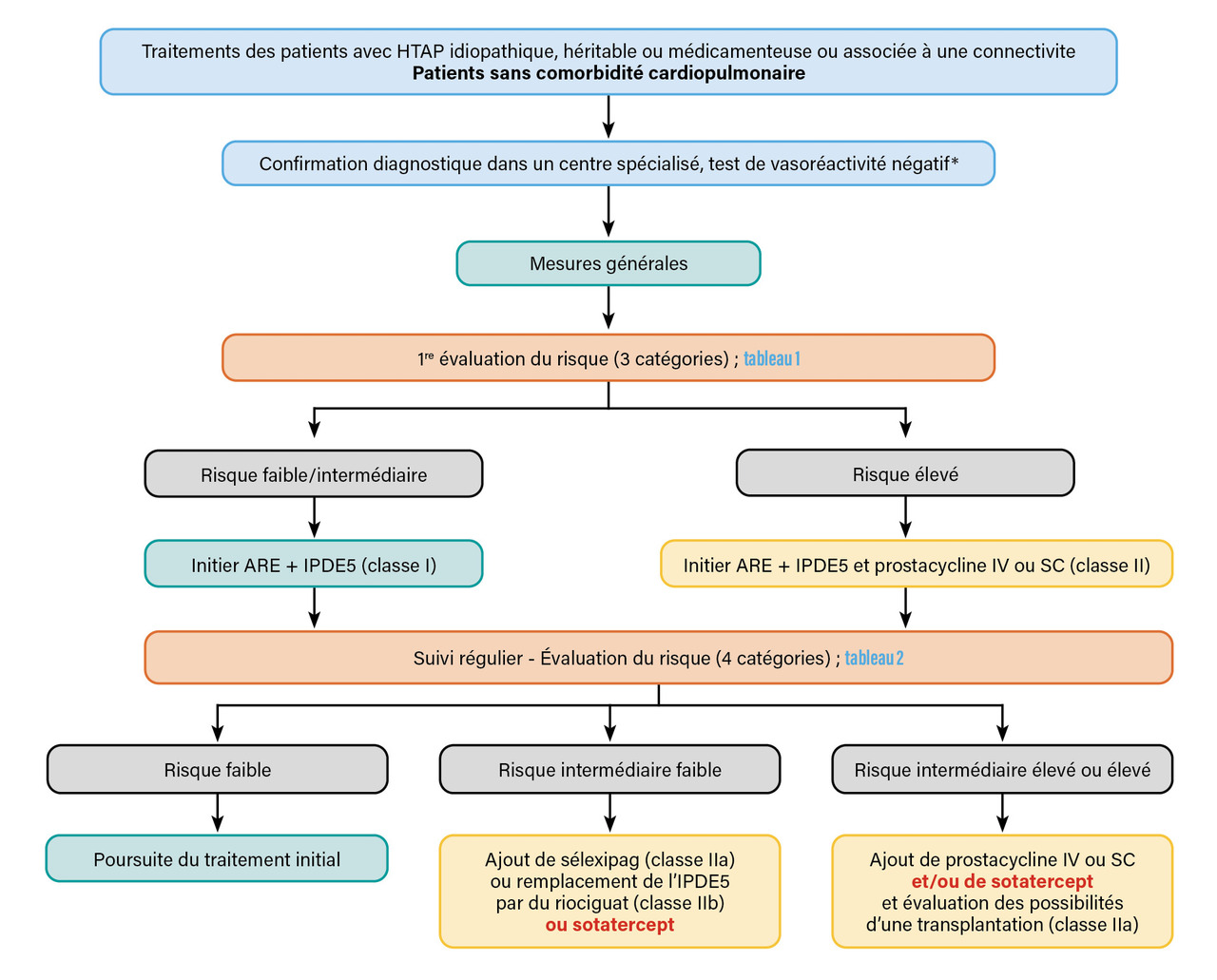
Un algorithme de traitement de l’HTAP est proposé dans les dernières recommandations communes des Sociétés européennes de cardiologie (European Society of Cardiology) et de pneumologie (European Respiratory Society). Cet algorithme, applicable dans l’HTAP idiopathique, héritable, médicamenteuse ou associée à une connectivite, est schématisé dans lafigure . Il distingue les patients sans comorbidités, pour lesquels cet algorithme s’applique principalement, des patients avec comorbidités cardiovasculaires (hypertension artérielle, diabète, obésité, maladie coronarienne) ou pulmonaires (fumeurs, capacité de diffusion du monoxyde de carbone [DLCO] basse), pour lesquels les recommandations thérapeutiques sont plus individuelles. Pour les malades sans comorbidités, l’algorithme est fondé sur l’évaluation du risque de mortalité à un an en utilisant la stratification du risque en trois niveaux au diagnostic et en quatre niveaux à la réévaluation (tableaux 1 et 2 ).1,17,18
Le risque de mortalité à un an est multiparamétrique, reposant sur des variables de sept ordres :
Chez les patients à risque faible ou intermédiaire, et sans comorbidités, il est recommandé de débuter par une association thérapeutique associant un ARE et un IPDE-5. Une réévaluation est entreprise entre trois et six mois après l’initiation du traitement, comportant notamment une réévaluation clinique (CF NYHA, signes d’insuffisance cardiaque droite), de la capacité d’exercice (TM6 et/ou épreuve d’effort cardiopulmonaire) et la mesure de biomarqueurs (BNP ou NT-proBNP). Une échocardiographie ainsi qu’un contrôle hémodynamique par cathétérisme cardiaque droit sont le plus souvent réalisés lors de cette première réévaluation.
En cas de réponse clinique jugée non satisfaisante, le traitement est modifié selon l’évaluation du risque en quatre strates (tableau 2) reposant sur les trois paramètres non invasifs que sont la CF NYHA, la distance parcourue au TM6 et un biomarqueur (BNP ou NT-proBNP).18
L’évaluation échocardiographique et surtout hémodynamique peut apporter des éléments complémentaires pour la décision thérapeutique.
Les objectifs thérapeutiques sont atteints si le patient est classé à risque faible à la première réévaluation. Si le patient est considéré à risque intermédiaire-faible, il est proposé d’associer à la bithérapie initiale un troisième médicament, le sélexipag (niveau de preuve et classe de recommandation IIa), ou bien de remplacer l’IPDE-5 par le riociguat (niveau de preuve et classe de recommandation IIb).
Les patients classés à risque intermédiaire-élevé ou à risque élevé lors de la réévaluation doivent bénéficier d’un traitement par prostacycline par voie intraveineuse ou sous-cutanée (en plus du traitement initial qui est poursuivi) et avoir une évaluation pour la transplantation pulmonaire s’ils sont éligibles.1
Un algorithme de traitement de l’HTAP est proposé dans les dernières recommandations communes des Sociétés européennes de cardiologie (European Society of Cardiology) et de pneumologie (European Respiratory Society). Cet algorithme, applicable dans l’HTAP idiopathique, héritable, médicamenteuse ou associée à une connectivite, est schématisé dans la
Le risque de mortalité à un an est multiparamétrique, reposant sur des variables de sept ordres :
- cliniques (CF NYHA, signes d’insuffisance cardiaque droite ou de progression des symptômes) ;
- fonctionnelles (distance parcourue au TM6) ;
- d’exercice (pic de consommation d’oxygène ou efficacité ventilatoire [VE/VCO2]) ;
- biologiques (brain natriuretic peptide [BNP] ou N-terminal-proBNP [NT-proBNP]) ;
- échocardiographiques (comme la taille de l’oreillette droite, la présence d’un épanchement péricardique ou le couplage ventriculo-artériel par la mesure du déplacement de l’anneau tricuspide vers l’apex (tricuspid annular plane systolic excursion [TAPSE]) rapportée à la pression artérielle pulmonaire systolique [PAPs]) ;
- radiologiques (IRM cardiaque) ;
- et hémodynamiques, mesurées lors du cathétérisme cardiaque droit initial (pression dans l’oreillette droite, index cardiaque, volume d’éjection systolique indexé [index cardiaque/fréquence cardiaque] et saturation veineuse en oxygène dans l’artère pulmonaire [SvO2]).
Chez les patients à risque faible ou intermédiaire, et sans comorbidités, il est recommandé de débuter par une association thérapeutique associant un ARE et un IPDE-5. Une réévaluation est entreprise entre trois et six mois après l’initiation du traitement, comportant notamment une réévaluation clinique (CF NYHA, signes d’insuffisance cardiaque droite), de la capacité d’exercice (TM6 et/ou épreuve d’effort cardiopulmonaire) et la mesure de biomarqueurs (BNP ou NT-proBNP). Une échocardiographie ainsi qu’un contrôle hémodynamique par cathétérisme cardiaque droit sont le plus souvent réalisés lors de cette première réévaluation.
En cas de réponse clinique jugée non satisfaisante, le traitement est modifié selon l’évaluation du risque en quatre strates (tableau 2) reposant sur les trois paramètres non invasifs que sont la CF NYHA, la distance parcourue au TM6 et un biomarqueur (BNP ou NT-proBNP).18
L’évaluation échocardiographique et surtout hémodynamique peut apporter des éléments complémentaires pour la décision thérapeutique.
Les objectifs thérapeutiques sont atteints si le patient est classé à risque faible à la première réévaluation. Si le patient est considéré à risque intermédiaire-faible, il est proposé d’associer à la bithérapie initiale un troisième médicament, le sélexipag (niveau de preuve et classe de recommandation IIa), ou bien de remplacer l’IPDE-5 par le riociguat (niveau de preuve et classe de recommandation IIb).
Les patients classés à risque intermédiaire-élevé ou à risque élevé lors de la réévaluation doivent bénéficier d’un traitement par prostacycline par voie intraveineuse ou sous-cutanée (en plus du traitement initial qui est poursuivi) et avoir une évaluation pour la transplantation pulmonaire s’ils sont éligibles.1
Lire aussi | Hypertension pulmonaire : les 10 messages-clés
Transplantation
La transplantation est à ce jour le seul traitement curatif de l’HTAP. Les patients qui restent à risque intermédiaire-élevé ou à risque élevé malgré un traitement médical maximal (en général une trithérapie comportant un analogue de la PGI2 par voie parentérale depuis au moins trois mois) sont candidats à la transplantation s’ils n’ont pas de contre-indication.
La transplantation bipulmonaire constitue désormais la technique de choix dans cette indication.19 Ce traitement n’est, bien entendu, envisageable que chez des patients relativement jeunes et ne présentant pas de comorbidités significatives.
La transplantation cardiopulmonaire reste indiquée en cas d’HTAP associée à une cardiopathie congénitale complexe non réparable pendant ou après transplantation pulmonaire.
En France, le pronostic des patients en attente de greffe s’est beaucoup amélioré avec les possibilités de demande d’accès prioritaire national à la transplantation auprès de l’Agence de la biomédecine (procédure de « superurgence »).20
La transplantation bipulmonaire constitue désormais la technique de choix dans cette indication.19 Ce traitement n’est, bien entendu, envisageable que chez des patients relativement jeunes et ne présentant pas de comorbidités significatives.
La transplantation cardiopulmonaire reste indiquée en cas d’HTAP associée à une cardiopathie congénitale complexe non réparable pendant ou après transplantation pulmonaire.
En France, le pronostic des patients en attente de greffe s’est beaucoup amélioré avec les possibilités de demande d’accès prioritaire national à la transplantation auprès de l’Agence de la biomédecine (procédure de « superurgence »).20
Perspectives et innovations thérapeutiques
De nouvelles molécules sont en cours de développement dans le cadre d’études de phases II et III, avec la perspective d’avancées thérapeutiques significatives dans les années à venir. Ces nouvelles molécules ciblent des voies physiopathologiques dysfonctionnelles, différentes de la dysfonction endothéliale.
Un des médicaments les plus prometteurs semble être le sotatercept, biothérapie qui agit sur la voie de signalisation du TGF-β (transforming growth factor) dont on sait qu’elle est très fortement impliquée dans la physiopathologie de l’HTAP. Le sotatercept est un piège à ligands qui a pour effet de restaurer l’équilibre entre la voie BMPR2 (antiproliférante ; bone morphogenetic protein receptor type 2) et la voie de l’activine (proproliférante) et ainsi de diminuer le remodelage vasculaire pulmonaire.
Les premières données d’efficacité de cette molécule dans l’HTAP sont très encourageantes, avec une amélioration des variables hémodynamiques, en particulier une diminution de la PAP et des RVP chez des malades déjà lourdement traités par bi-ou trithérapie, mais également des variables cliniques (CF NYHA), fonctionnelles (distance parcourue au TM6) et biologiques (NT-proBNP).21,22
D’autres traitements, comme des inhibiteurs de tyrosine kinase par voie inhalée, qui ciblent la voie du PDGF (platelet derived growth factor), semblent également prometteurs avec une étude de phase II ayant montré un effet bénéfique du séralutinib sur l’hémodynamique. De nombreuses autres molécules sont également en cours d’étude, ciblant la dysfonction endothéliale (stimulateur de la guanylate cyclase par voie inhalée) ou d’autres voies physiopathologiques dysfonctionnelles.
Un des médicaments les plus prometteurs semble être le sotatercept, biothérapie qui agit sur la voie de signalisation du TGF-β (transforming growth factor) dont on sait qu’elle est très fortement impliquée dans la physiopathologie de l’HTAP. Le sotatercept est un piège à ligands qui a pour effet de restaurer l’équilibre entre la voie BMPR2 (antiproliférante ; bone morphogenetic protein receptor type 2) et la voie de l’activine (proproliférante) et ainsi de diminuer le remodelage vasculaire pulmonaire.
Les premières données d’efficacité de cette molécule dans l’HTAP sont très encourageantes, avec une amélioration des variables hémodynamiques, en particulier une diminution de la PAP et des RVP chez des malades déjà lourdement traités par bi-ou trithérapie, mais également des variables cliniques (CF NYHA), fonctionnelles (distance parcourue au TM6) et biologiques (NT-proBNP).21,22
D’autres traitements, comme des inhibiteurs de tyrosine kinase par voie inhalée, qui ciblent la voie du PDGF (platelet derived growth factor), semblent également prometteurs avec une étude de phase II ayant montré un effet bénéfique du séralutinib sur l’hémodynamique. De nombreuses autres molécules sont également en cours d’étude, ciblant la dysfonction endothéliale (stimulateur de la guanylate cyclase par voie inhalée) ou d’autres voies physiopathologiques dysfonctionnelles.
Lire aussi | Dépistage et diagnostic de l’hypertension pulmonaire
Propositions à la suite du 7e Congrès mondial de l’HTP
La place du sotatercept dans l’algorithme de prise en charge de l’HTAP a été rediscutée lors du symposium mondial sur l’HTP de juin 2024,23 à la suite des résultats positifs des études de phases II et III (PULSAR et STELLAR).21,22 Le sotatercept apparaît donc dans l’algorithme thérapeutique de l’HTAP et peut être discuté dès la première réévaluation chez les patients qui ne sont pas à faible risque.
Ainsi, il peut être proposé en alternative au selexipag ou au riociguat à un patient à risque intermédiaire faible. Chez les patients restant à risque intermédiaire-élevé ou à haut risque à la réévaluation, le sotatercept peut être proposé en alternative à l’ajout d’une prostacycline par voie parentérale.
Enfin, une quadrithérapie est possible pour les patients les plus sévères qui doivent en parallèle être évalués pour la transplantation pulmonaire.
Néanmoins, actuellement, en France, le sotatercept n’est disponible qu’en accès précoce chez les patients avec HTAP qui restent en classe fonctionnelle II ou III sur l’échelle NYHA malgré une trithérapie incluant une prostacycline par voie parentérale. Il peut être également proposé dans le cadre d’essais cliniques.
Enfin, la présence de comorbidités cardiopulmonaires doit rendre le clinicien vigilant lors de l’instauration du traitement spécifique de l’hypertension artérielle pulmonaire.
Ainsi, il peut être proposé en alternative au selexipag ou au riociguat à un patient à risque intermédiaire faible. Chez les patients restant à risque intermédiaire-élevé ou à haut risque à la réévaluation, le sotatercept peut être proposé en alternative à l’ajout d’une prostacycline par voie parentérale.
Enfin, une quadrithérapie est possible pour les patients les plus sévères qui doivent en parallèle être évalués pour la transplantation pulmonaire.
Néanmoins, actuellement, en France, le sotatercept n’est disponible qu’en accès précoce chez les patients avec HTAP qui restent en classe fonctionnelle II ou III sur l’échelle NYHA malgré une trithérapie incluant une prostacycline par voie parentérale. Il peut être également proposé dans le cadre d’essais cliniques.
Enfin, la présence de comorbidités cardiopulmonaires doit rendre le clinicien vigilant lors de l’instauration du traitement spécifique de l’hypertension artérielle pulmonaire.
Références
1. Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 2022;2200879.
2. Grünig E, MacKenzie A, Peacock AJ, et al. Standardized exercise training is feasible, safe, and effective in pulmonary arterial and chronic thromboembolic pulmonary hypertension: Results from a large European multicentre randomized controlled trial. Eur Heart J 2021;42(23):2284‑95.
3. Preston IR, Roberts KE, Miller DP, et al. Effect of warfarin treatment on survival of patients with pulmonary arterial hypertension (PAH) in the registry to evaluate early and long-term PAH disease management (REVEAL). Circulation 2015;132(25):2403‑11.
4. Sitbon O, Humbert M, Jais X, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005;111(23):3105‑11.
5. McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: The impact of epoprostenol therapy. Circulation 2002;106(12):1477‑82.
6. Boucly A, Savale L, Jaïs X, et al. Association between initial treatment strategy and long-term survival in pulmonary arterial hypertension. Am J Respir Crit Care Med 2021;204(7):842-54.
7. Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 1996;334(5):296‑301.
8. Simonneau G, Barst RJ, Galie N, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: A double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med 2002;165(6):800‑4.
9. Sitbon O, Channick R, Chin KM, et al. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med 2015;373(26):2522‑33.
10. Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002;346(12):896‑903.
11. Galie N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: Results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008;117(23):3010‑9.
12. Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013;369(9):809‑18.
13. Galie N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 2005;353(20):2148‑57.
14. Galie N, Brundage BH, Ghofrani HA, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation 2009;119(22):2894‑903.
15. Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 2013;369(4):330‑40.
16. Ghofrani HA, D’Armini AM, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med 2013;369(4):319‑29.
17. Boucly A, Weatherald J, Savale L, et al. Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. Eur Respir J 2017;50(2):1700889.
18. Boucly A, Weatherald J, Savale L, et al. External validation of a refined four-stratum risk assessment score from the French pulmonary hypertension registry. Eur Respir J 2022;59(6):2102419.
19. Hoeper MM, Benza RL, Corris P, et al. Intensive care, right ventricular support and lung transplantation in patients with pulmonary hypertension. Eur Respir J 2019;53(1):1801906.
20. Savale L, Le Pavec J, Mercier O, et al. Impact of high-priority allocation on lung and heart-lung transplantation for pulmonary hypertension. Ann Thorac Surg 2017;104(2):404‑11.
21. Humbert M, McLaughlin V, Gibbs JSR, et al. Sotatercept for the treatment of pulmonary arterial hypertension. N Engl J Med 2021;384(13):1204‑15.
22. Hoeper MM, Badesch DB, Ghofrani HA, et al. Phase 3 trial of sotatercept for treatment of pulmonary arterial hypertension. N Engl J Med 2023;388(16):1478‑90.
23. Chin KM, Gaine SP, Gerges C, et al. Treatment algorithm for pulmonary arterial hypertension. Eur Respir J 2024;2401325.
2. Grünig E, MacKenzie A, Peacock AJ, et al. Standardized exercise training is feasible, safe, and effective in pulmonary arterial and chronic thromboembolic pulmonary hypertension: Results from a large European multicentre randomized controlled trial. Eur Heart J 2021;42(23):2284‑95.
3. Preston IR, Roberts KE, Miller DP, et al. Effect of warfarin treatment on survival of patients with pulmonary arterial hypertension (PAH) in the registry to evaluate early and long-term PAH disease management (REVEAL). Circulation 2015;132(25):2403‑11.
4. Sitbon O, Humbert M, Jais X, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005;111(23):3105‑11.
5. McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: The impact of epoprostenol therapy. Circulation 2002;106(12):1477‑82.
6. Boucly A, Savale L, Jaïs X, et al. Association between initial treatment strategy and long-term survival in pulmonary arterial hypertension. Am J Respir Crit Care Med 2021;204(7):842-54.
7. Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 1996;334(5):296‑301.
8. Simonneau G, Barst RJ, Galie N, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: A double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med 2002;165(6):800‑4.
9. Sitbon O, Channick R, Chin KM, et al. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med 2015;373(26):2522‑33.
10. Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002;346(12):896‑903.
11. Galie N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: Results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008;117(23):3010‑9.
12. Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013;369(9):809‑18.
13. Galie N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 2005;353(20):2148‑57.
14. Galie N, Brundage BH, Ghofrani HA, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation 2009;119(22):2894‑903.
15. Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 2013;369(4):330‑40.
16. Ghofrani HA, D’Armini AM, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med 2013;369(4):319‑29.
17. Boucly A, Weatherald J, Savale L, et al. Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. Eur Respir J 2017;50(2):1700889.
18. Boucly A, Weatherald J, Savale L, et al. External validation of a refined four-stratum risk assessment score from the French pulmonary hypertension registry. Eur Respir J 2022;59(6):2102419.
19. Hoeper MM, Benza RL, Corris P, et al. Intensive care, right ventricular support and lung transplantation in patients with pulmonary hypertension. Eur Respir J 2019;53(1):1801906.
20. Savale L, Le Pavec J, Mercier O, et al. Impact of high-priority allocation on lung and heart-lung transplantation for pulmonary hypertension. Ann Thorac Surg 2017;104(2):404‑11.
21. Humbert M, McLaughlin V, Gibbs JSR, et al. Sotatercept for the treatment of pulmonary arterial hypertension. N Engl J Med 2021;384(13):1204‑15.
22. Hoeper MM, Badesch DB, Ghofrani HA, et al. Phase 3 trial of sotatercept for treatment of pulmonary arterial hypertension. N Engl J Med 2023;388(16):1478‑90.
23. Chin KM, Gaine SP, Gerges C, et al. Treatment algorithm for pulmonary arterial hypertension. Eur Respir J 2024;2401325.
Dans cet article

Résumé
La prise en charge de l’hypertension artérielle pulmonaire (HTAP) s’est considérablement améliorée au cours des dernières années, parallèlement aux progrès réalisés dans la compréhension des mécanismes physiopathologiques de la maladie, au développement de médicaments innovants et à l’application de stratégies thérapeutiques plus agressives. L’HTAP étant une maladie rare, sa prise en charge est assurée dans un centre d’expertise en hypertension pulmonaire (HTP). Elle associe des mesures générales, des traitements symptomatiques et des thérapeutiques dites « spécifiques » ciblant l’équilibre vasoconstriction/vasodilatation et proprolifération/antiprolifération des artères pulmonaires. Malgré les progrès thérapeutiques importants de ces dernières années, le pronostic de l’HTAP reste sombre et la transplantation bipulmonaire (ou cardiopulmonaire) qui est proposée aux patients les plus sévèrement atteints constitue à ce jour le seul traitement curatif.