ConnaÎtre les principales hypothèses diagnostiques et les examens complémentaires pertinents.
Apprécier la gravité d’une anémie.
ConnaÎtre les urgences liées à l’anémie et les signes de gravité (terrain, rapidité d’installation et profondeur).
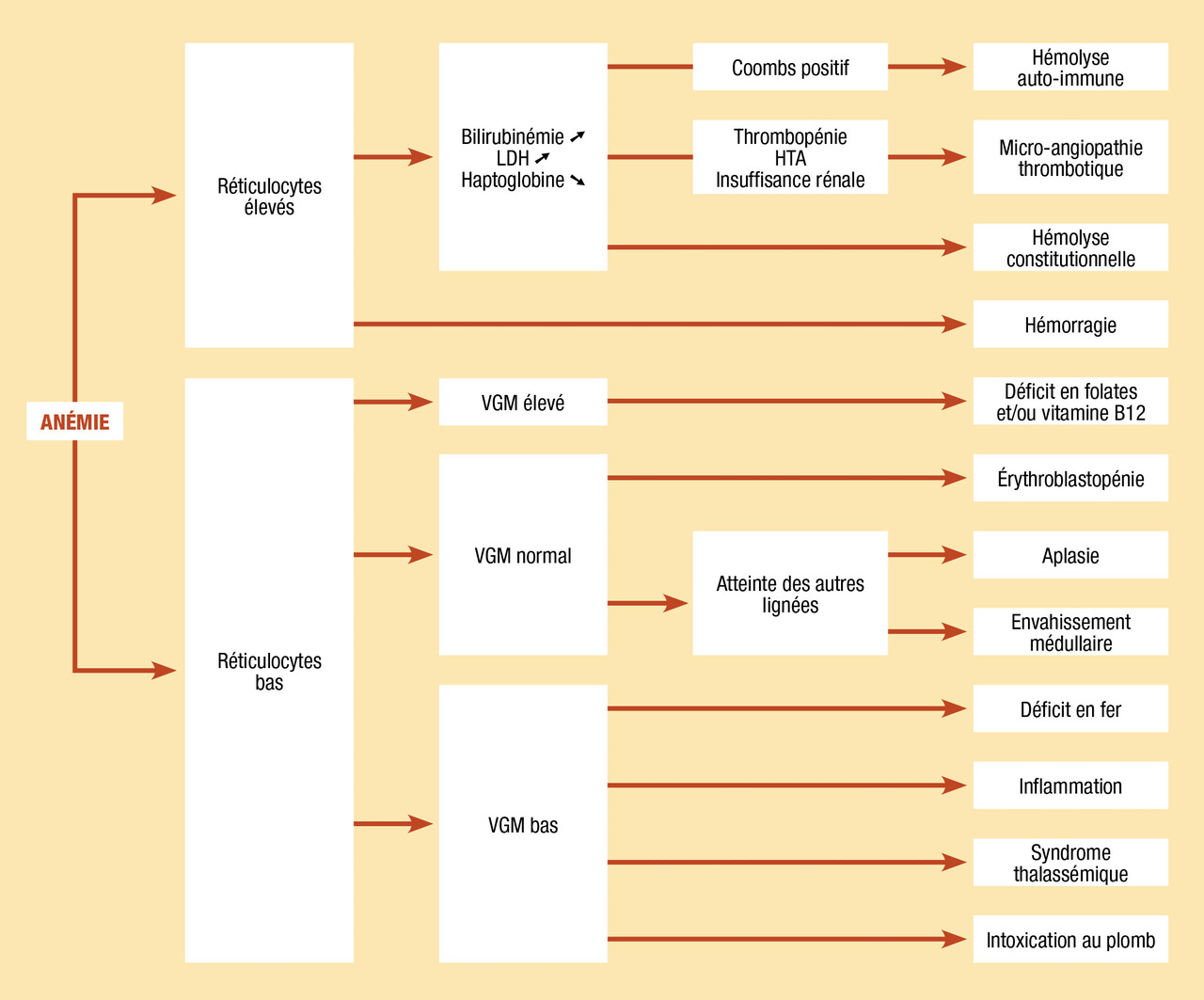
Outils diagnostiques
Anémies microcytaires
Carence en fer
- carence d’apports soit par carence maternelle, soit par régime lacté prolongé et/ou pauvre en fer dans le cadre d’un régime inapproprié : enquête alimentaire ;
- perte de fer par hémorragie chronique (à laquelle s’apparente la spoliation sanguine par prélèvements sanguins répétés et/ou de volume excessif sur un organisme déjà anémié) : recherche de sang dans les selles ;
- malabsorption secondaire à différentes pathologies digestives acquises ou constitutionnelles : troubles du transit, signes cliniques de malabsorption. À noter que le « pica » (ingestion de substances non alimentaires) est répandu dans certaines cultures et peut induire une malabsorption du fer, notamment par consommation d’argile.
Anémie inflammatoire
Syndrome thalassémique
Autres causes rares d’anémie microcytaire
Anémies normo- ou macrocytaires
Il importe toutefois de tenir compte d’un délai de quelques jours d’apparition des réticulocytes si l’anémie est récente. En présence d’une hémoglobine basse, la régénération peut être considérée comme franchement insuffisante si elle est inférieure à 50 000/mm3 (50 G/L), bien qu’il n’y ait pas de seuil formel pour apprécier le caractère arégénératif d’une anémie. Une fois écartée une insuffisance rénale avancée et une hypothyroïdie, le caractère arégénératif d’une anémie évoque une cause centrale et amène, sauf exception, à la réalisation d’un myélogramme.
Anémie arégénérative
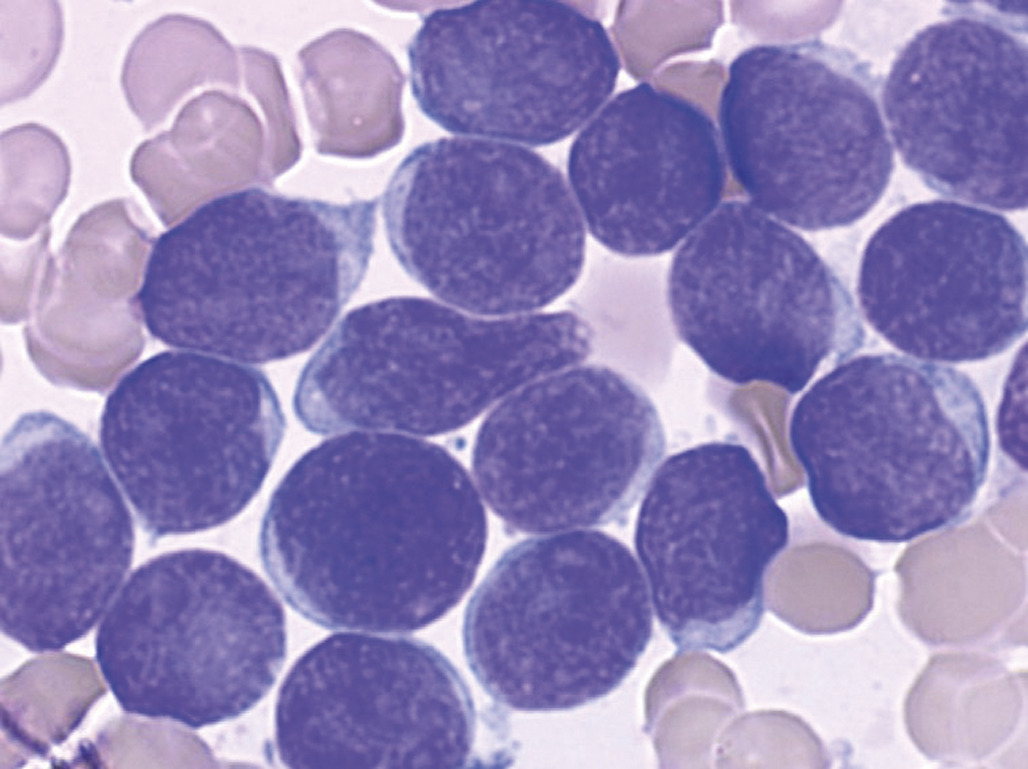
Envahissement médullaire
Une anémie associée à une altération de l’état général, un syndrome tumoral, des douleurs osseuses sont évocateurs d’un envahissement médullaire (Dans un contexte plus altéré (fièvre, anomalie de l’hémostase avec hypofibrinémie, hypertriglycéridémie, hyperferritinémie), l’infiltration de la moelle par des lymphocytes T activés induit un syndrome d’activation macrophagique (« SAM »), plus justement appelé syndrome d’activation lymphohistiocytaire (
Carence en vitamine B9 (folates) et/ou B12 (cobalamine)
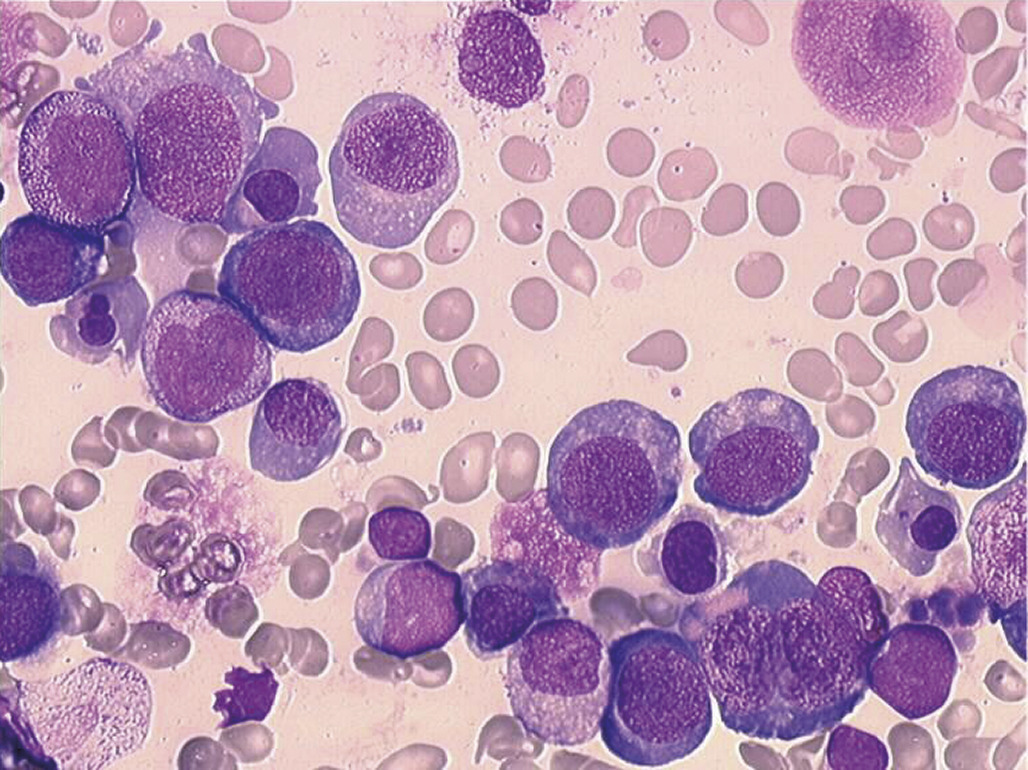
Les vitamines B9 et B12 sont impliquées dans la synthèse des bases nucléiques. Le déficit dans l’une ou l’autre de ces vitamines va altérer globalement l’hématopoïèse, mais l’atteinte de la lignée érythropoïétique est la plus marquée. La macrocytose s’accompagne d’une mégaloblastose (Le déficit en vitamine B9 et/ou B12 s’observe essentiellement dans des situations de carence constitutionnelle et/ou de régime alimentaire inapproprié. Des signes neurologiques (retard psychomoteur, hypotonie, dyskinésie) sont possibles en cas de carence profonde. Les déficits spécifiques d’absorption de la vitamine B12 par déficit congénital ou acquis en facteur intrinsèque sont exceptionnels chez l’enfant. Biologiquement, une neutropénie, une thrombopénie à plaquettes géantes sont associées à l’anémie. Le myélogramme confirme la mégaloblastose avec un asynchronisme de maturation nucléocytoplasmique.
Érythroblastopénie sélective
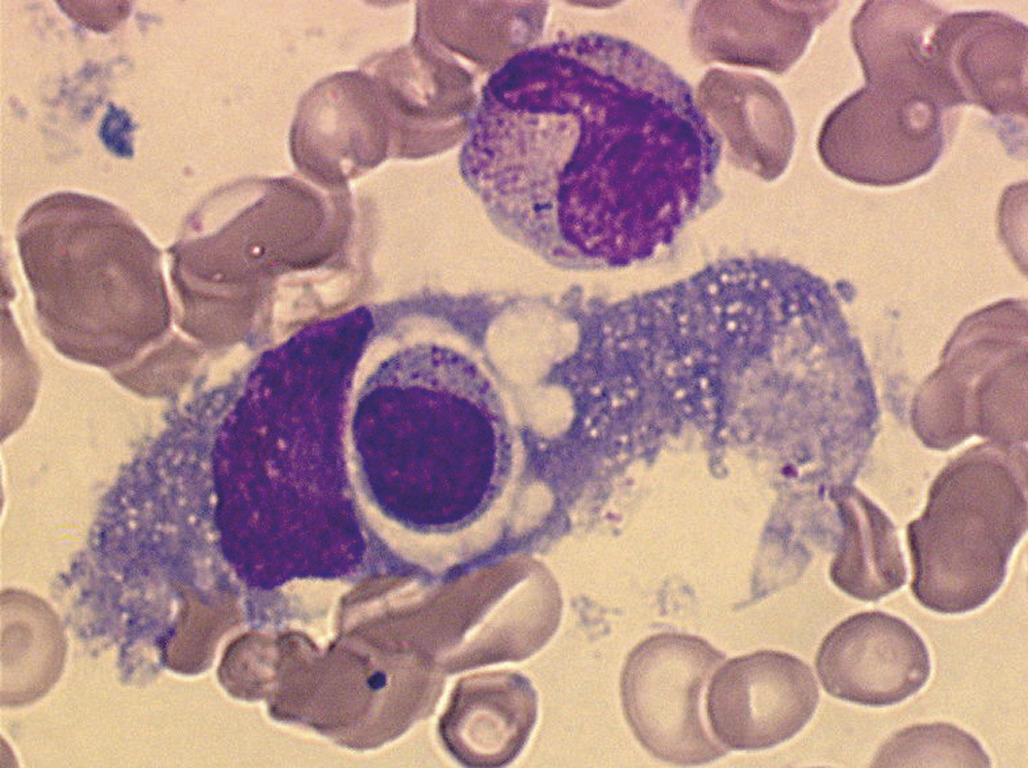
Le parvovirus B19, qui altère spécifiquement le précurseur érythropoïétique et bloque la production d’érythrocytes pendant quelques jours, induit une érythroblastopénie. En l’absence d’une pathologie du globule rouge sous-jacente, cette altération transitoire de l’érythropoïèse passe inaperçue. Dans un contexte d’anémie hémolytique chronique (sphérocytose, drépanocytose ; v. infra), l’arrêt transitoire de l’érythropoïèse s’accompagne alors d’une anémie arégénérative qui peut être profonde. L’infection à parvovirus B19 peut être révélatrice de cette pathologie hémolytique sous-jacente. La fièvre, l’éruption qui précède l’anémie peuvent passer inaperçues. L’anémie est normocytaire, le taux de réticulocytes est très bas, inférieur à 5 000/mm3. Sur le frottis médullaire, on peut visualiser des proérythroblastes géants avec une chromatine dense non mottée et de volumineux nucléoles. (Chez l’enfant entre 1 et 4 ans, une anémie liée à une érythroblastopénie transitoire, non liée au parvovirus B19, peut être observée. Le blocage est plus prolongé que lors de l’infection à parvovirus B19. L’évolution est spontanément réversible bien que pouvant nécessiter un support transfusionnel ponctuel. Elle est probablement d’origine virale, mais non identifiée à ce jour.
Enfin, la rare maladie de Blackfan-Diamond est une érythroblastopénie constitutionnelle liée à une pathologie ribosomale. Elle se présente comme une anémie néonatale profondément arégénérative avec au myélogramme une altération sélective de la lignée érythroblastique, sans infection à parvovirus B19. La moitié des enfants atteints ont des malformations associées (syndrome de Pierre Robin, malformation urogénitale notamment).
Altération globale de l’hématopoïèse : aplasie médullaire
Une anémie isolée peut être le premier signe d’une évolution vers une dégradation globale de l’hématopoïèse bien que le diagnostic initial soit fait le plus souvent sur une atteinte de deux ou des trois lignées. L’aplasie médullaire peut être d’origine immunologique, notamment lorsqu’elle suit un diagnostic d’hépatite auto-immune. Elle peut être aussi constitutionnelle, associée ou non à d’autres signes cliniques tels que les anomalies osseuses. La maladie de Fanconi est la plus fréquente de ces maladies rares. Dans les deux tiers des cas, les premiers symptômes sont des malformations congénitales (squelettiques, cutanées, urogénitales, anomalies des membres uni- ou bilatérales, microcéphalie et/ou microphtalmie), mais l’anémie souvent macrocytaire peut être le premier signe d’évolution progressive vers l’aplasie. Même si le phénotype clinique permet le diagnostic confirmé par la génétique, le myélogramme est nécessaire pour apprécier le degré d’altération médullaire et évaluer son évolutivité. La maladie de Fanconi est liée à une altération d’un des gènes d’une large famille de gènes impliqués dans la réparation de l’ADN. De nombreuses autres étiologies moléculaires d’aplasie médullaire sont désormais identifiées, syndromiques ou non.Anémies régénératives
Hémolyse constitutionnelle membranaire
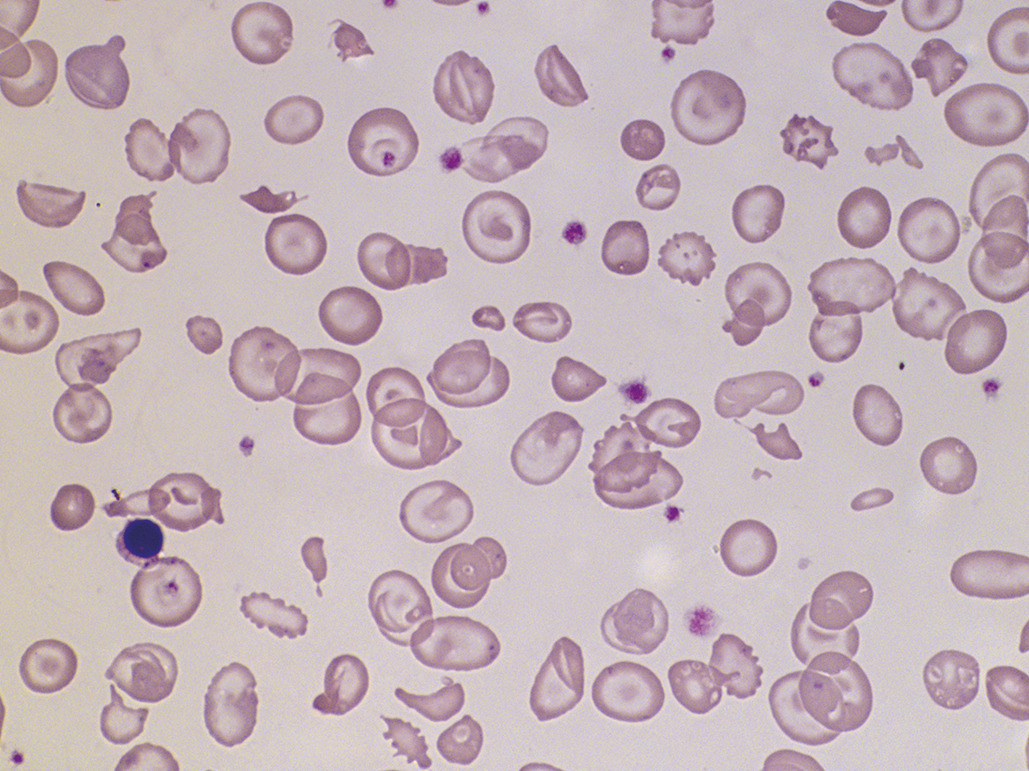
La forme la plus fréquente est la sphérocytose (Hémolyse constitutionnelle enzymatique
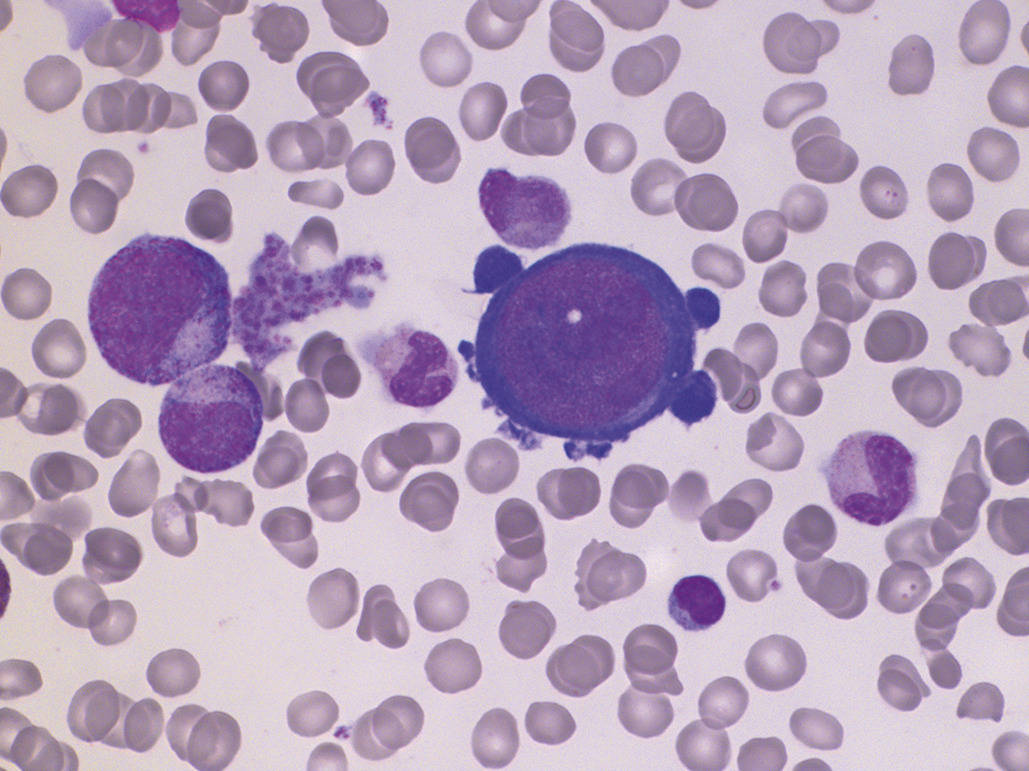
Le déficit en glucose-6-phosphate déshydrogénase (G6PD) est le plus fréquent des déficits enzymatiques associés à une hémolyse. Il touche principalement les garçons, mais les filles peuvent être atteintes. Le déficit est le plus souvent totalement asymptomatique, mais il peut aussi évoluer par poussée d’hémolyse déclenchée par certains traitements, infections ou alimentation à base de fèves. Le diagnostic se fait sur le dosage enzymatique, on peut distinguer au frottis sanguin la présence d’hemighosts (Hémolyse par hémoglobinopathie
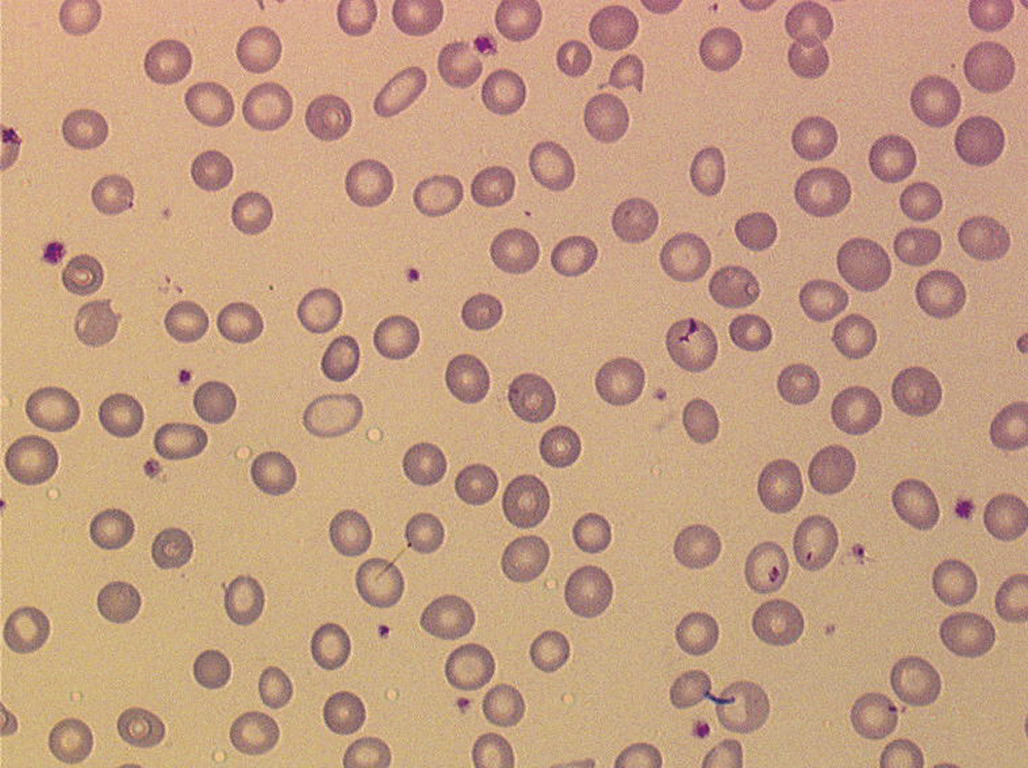
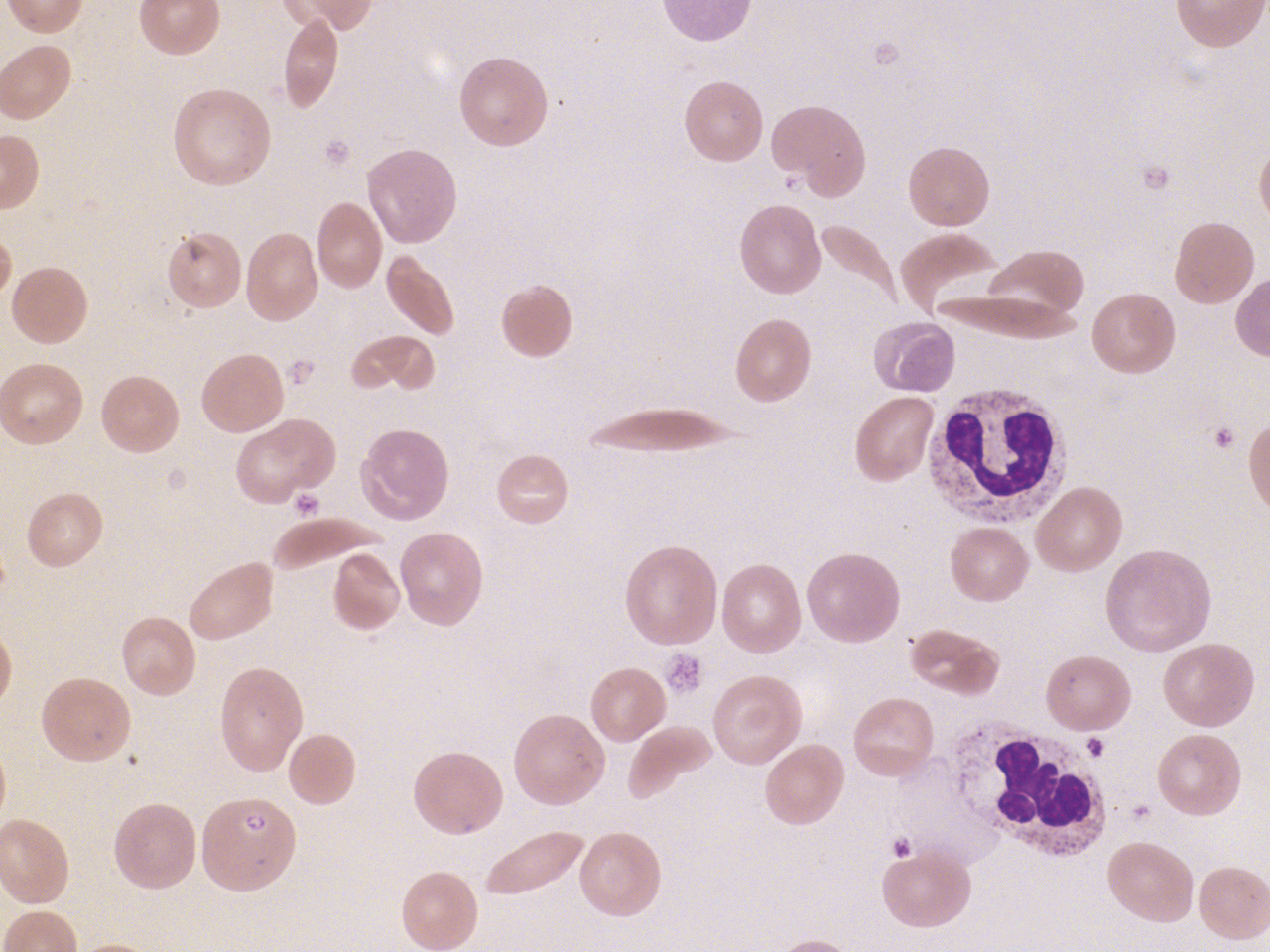
Une anémie hémolytique chez un enfant dont la famille est d’origine africaine ou antillaise fait évoquer une drépanocytose, maladie génétique de l’hémoglobine due à une mutation sur le gène de la chaîne b-globine. L’hémoglobine S (HbS), produit de cette mutation, se polymérise et fragilise l’érythrocyte, rendant compte de l’anémie hémolytique. L’anémie est normocytaire, associée à une réticulocytose, une hyperbilirubinémie, un taux de LDH élevé, une haptoglobine basse ; le frottis retrouve des hématies en faucille (L’augmentation de la viscosité sanguine, la rigidification des érythrocytes, et leur adhésion à l’endothélium vasculaire induisent les complications vasculaires de la maladie, qui peuvent être aussi révélatrices (« crise vaso-occlusive » douloureuse ou syndrome thoracique aigu). Pour les enfants nés en France, le diag- nostic est fait grâce au dépistage systématique en période néonatale. Outre les échecs « logistiques » du dépistage, celui-ci reste ciblé sur l’origine géographique des parents et peut être pris en défaut.
Hémolyses auto-immunes
Les anémies hémolytiques auto-immunes sont induites par la fixation d’un anticorps à la surface des globules rouges entraînant une destruction intravasculaire. L’hémolyse peut être intense. L’anémie est normo- ou macrocytaire, associée à une hyperbilirubinémie, un taux de LDH élevé et une haptoglobine basse ou nulle. Le test de Coombs identifie les anticorps, qui peuvent être dits « chauds » (fixation à 37 °C) de type immunoglobuline G (IgG) fixant ou non le complément ou « froids » (fixation inférieure à 37 °C, optimale à 22 °C ou plus bas), le plus souvent alors IgM, parfois IgG. Dans les deux cas, le processus peut être isolé ou associé à une thrombopénie auto-immune (syndrome d’Evans). Il faudra dans tous les cas chercher un éventuel déficit immunitaire primitif à l’origine de cette anémie hémolytique. Entre 10 et 15 % des anémies hémolytiques sont d’origine infectieuse, le germe le plus classiquement évoqué étant le mycoplasme. Une réticulocytose basse peut s’observer quand l’hémolyse inclut les progéniteurs érythroblastiques. La surveillance évolutive d’un enfant chez qui une anémie hémolytique auto-immune a été identifiée doit être très attentive : l’hémolyse peut conduire à une menace vitale anémique en quelques heures.Hémolyse micro-angiopathique
L’anémie s’inscrit dans un contexte pathologique complexe classiquement lié à une diarrhée sanglante avec insuffisance rénale, protéinurie, hypertension artérielle, parfois atteinte neurologique. L’anémie est hémolytique, mais se caractérise par la présence de schizocytes (Hémolyses infectieuses
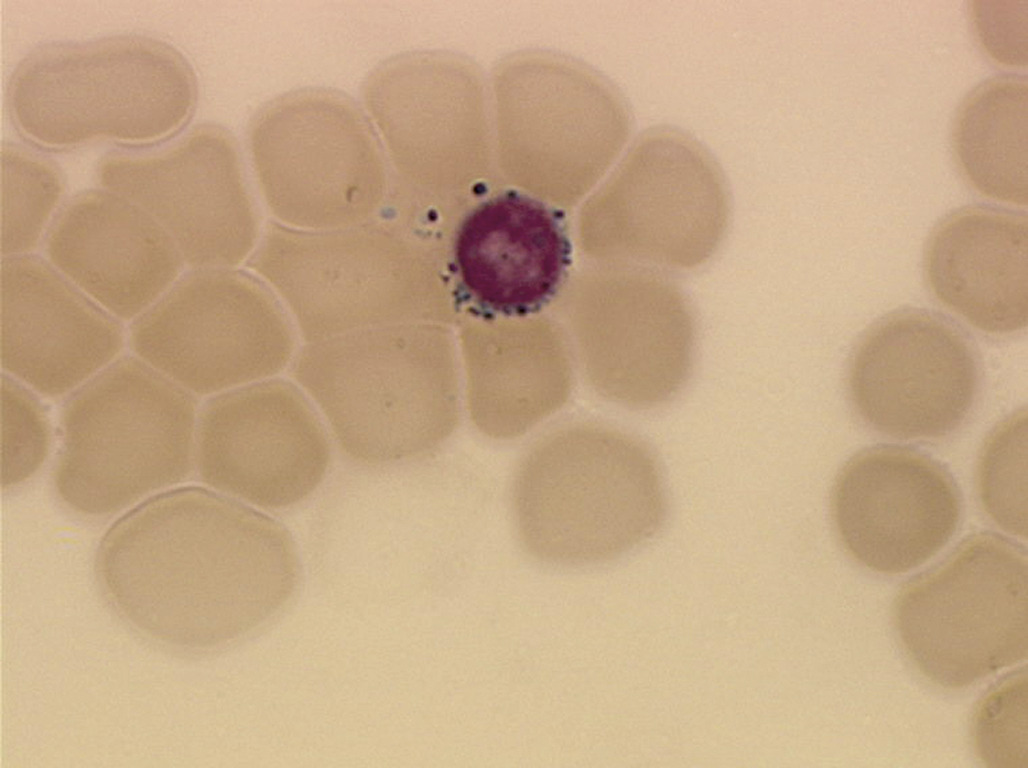
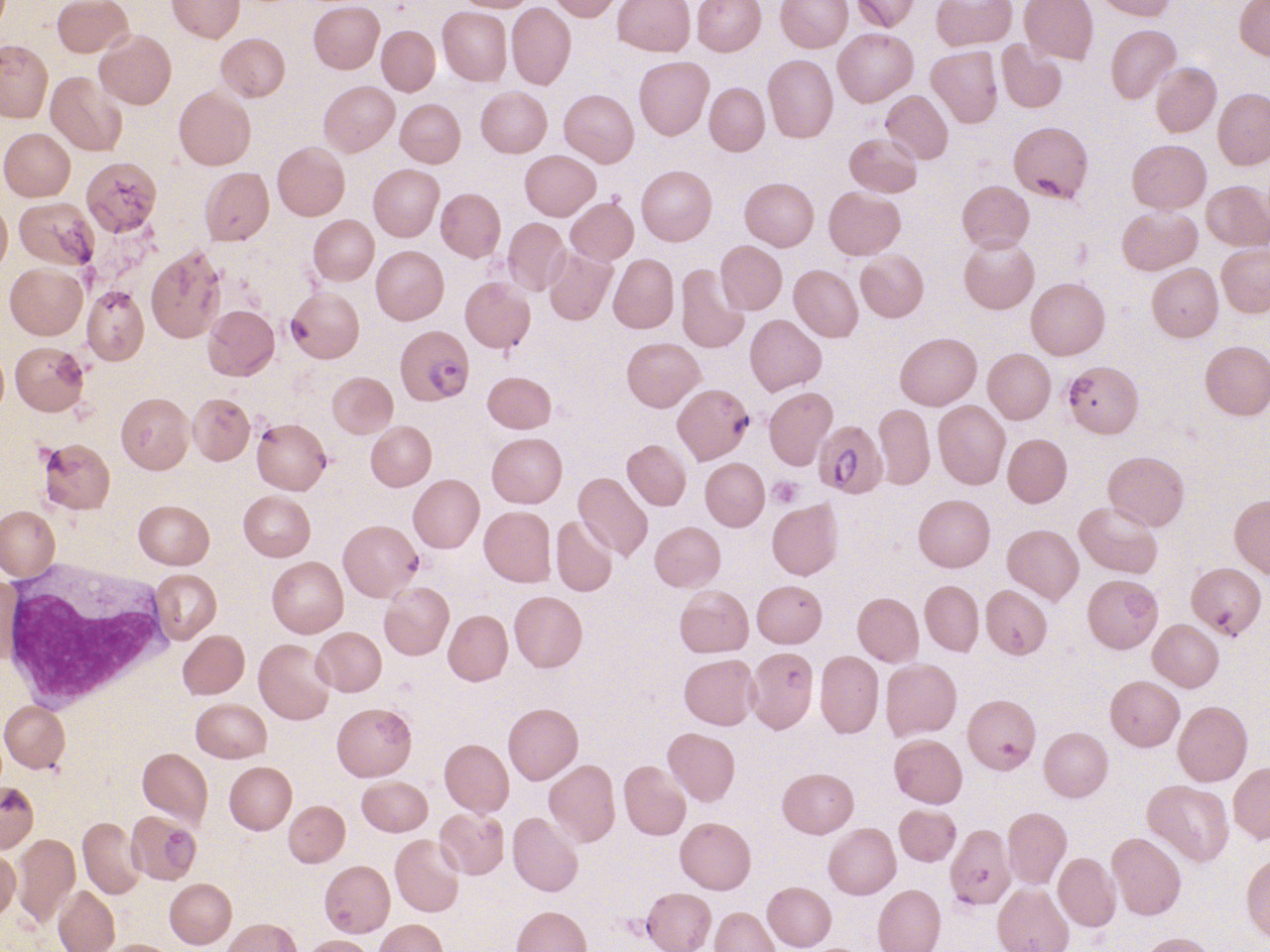
Le paludisme est une cause classique d’hémolyse plus ou moins sévère. Le contexte (fièvre en retour de zone d’épidémie) est le plus souvent évocateur, mais des cas de paludisme d’importation sont observés. Le diagnostic est fait sur le frottis et la goutte épaisse. Au frottis, on visualise à l’intérieur des érythrocytes le parasite donnant un aspect de « bagues à chaton » (D’autres infections peuvent être à l’origine d’une hémolyse, mais par des mécanismes indirects déjà cités (auto-anticorps sur infection à mycoplasme ou micro-angiopathie par infection à Escherichia coli).
Hémolyse médicamenteuse
Indépendamment du déficit en G6PD, plus de 130 médicaments sont susceptibles d’induire une anémie hémolytique immunologique ; 40 % sont des antibiotiques, pénicilline et céphalosporine essentiellement. L’anticorps est dirigé contre le médicament, et sa liaison avec l’hématie induit l’hémolyse. Il n’y a pas d’influence de la dose, de la voie d’administration ni de la durée. Le diagnostic est le plus souvent présomptif, après exclusion des autres cas d’hémolyse.L’anémie est donc un motif très fréquent de consultation en pédiatrie, à travers des étiologies extrêmement variables et parfois intriquées. L’algorithme de classification (
POINTS FORTS À RETENIR
L’anémie est une pathologie fréquente définie par une baisse de l’hémoglobine en dessous de seuils qui sont variables selon l’âge.
L’identification des caractéristiques biologiques « basiques » de l’anémie (volume globulaire moyen, taux de réticulocytes, bilirubinémie essentiellement) est cruciale pour la compréhension du mécanisme, mais le contexte, la rapidité de l’apparition du syndrome anémique et l’examen clinique sont indispensables à intégrer dans la démarche diagnostique.
La carence martiale est la cause la plus fréquente en pays industrialisés.
Devant une anémie normo- ou macrocytaire arégénérative, il faut – sauf rares exceptions – compléter le bilan par un myélogramme.
Les anémies normo- ou macrocytaires régénératives à bilirubine élevée font rechercher les différents processus d’hémolyse. L’association à une thrombopénie doit faire rechercher en urgence une micro-angiopathie thrombotique.
Une anémie hémolytique auto-immune peut conduire en quelques heures à une anémie profonde et à une menace vitale.
Prise en charge de l’anémie par carence martiale
Physiopathologie
Fer : élément constitutif de l’hème, une fois associé aux molécules de globines, ils forment l’hémoglobine.
Hémoglobine : 80 % du fer de l’organisme. Sa production nécessite 20 fois plus de fer que la quantité apportée par l’alimentation, d’où le recyclage par les macrophages à partir des globules rouges sénescents.
Protéines du fer :
– ferritine : forme de stockage du fer ;
– transferrine : protéine de transport.
En cas de carence martiale :
– diminution de la ferritine avec épuisement des réserves ;
– puis diminution du taux sérique de fer ;
– et augmentation du coefficient de saturation de la transferrine (rapport fer/transferrine) ;
– quand le fer délivré aux érythrocytes devient insuffisant pour l’érythropoïèse, on constate une diminution progressive de la synthèse de l’hémoglobine ;
– contenu en hémoglobine diminuée dans chacune des formes des érythroblastes alors que les divisions cellulaires sont maintenues ;
– érythrocytes contenant de moins en moins d’hémoglobine (hypochromie) et étant de plus en plus petits (microcytose).
Clinique
Anémie chronique, d’installation progressive.
Présence d’un syndrome carentiel : fragilité des phanères (chute des cheveux, ongles cassants), fragilité des muqueuses (glossite, œsophagite).
Hémogramme :
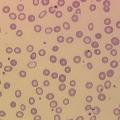
• anémie microcytaire (VGM abaissé selon l’âge), hypochrome (concentration corpusculaire moyenne en hémoglobine [CCMH] < 31 %) ;
• association possible à une thrombocytose réactionnelle :
– frottis sanguin : anisocytose,
– bilan martial,
– dosage uniquement de la ferritine (abaissée en cas de carence martiale, sauf si syndrome inflammatoire associé),
– ou en association avec celui du fer sérique abaissé < 11 µmol/L avec un des critères suivants :
° transferrine augmentée,
° coefficient de saturation de la transferrine diminué,
° capacité totale de fixation de la transferrine augmentée.
Après affirmation de la carence martiale, l’enjeu est d’en définir l’étiologie. Il existe 3 grands mécanismes de carence martiale :
– carence d’apports : cause la plus fréquente. La plupart du temps, il s’agit soit d’une carence maternelle, soit d’un régime lacté prolongé et/ou pauvre en fer, soit d’une erreur nutritionnelle (lait de vache avant 2 ans, lait d’amande) ;
– pertes de fer par saignement chronique : œsophagite sur reflux gastro-œsophagien, diverticule de Meckel, troubles de l’hémostase, parasitoses intestinales, exceptionnelle hémosidérose pulmonaire ;
– malabsorption : maladie cœliaque, autres diarrhées chroniques, « Pica » par ingestion de terre.
Traitement de la cause.
Traitement curatif : par voie orale : 5 à 10 mg/kg de sels ferreux. Fractionnée en 3 prises à distance des repas. Les parents doivent être informés des possibles effets indésirables tels que coloration des selles en noir, troubles digestifs (douleurs abdominales, diarrhées ou constipation), céphalées (rares). La durée de traitement recommandée est de 3 à 6 mois (jusqu’à normalisation de la ferritine).
La réponse au traitement est rapide, avec la survenue d’une élévation des réticulocytes (« crise réticulocytaire ») vers le dixième jour.
Traitement préventif : 500 mL d’équivalent de lait (lait maternel ou préparation de suite) chez tout nouveau-né ou nourrisson.
Le traitement parentéral doit être réservé aux très rares cas où un traitement per os bien conduit s'avère impossible ou inefficace. Sauf défaut d’observance manifeste, l’inefficacité du traitement per os doit faire rechercher une anomalie d’absorption et/ou de métabolisme du fer. L’injection parentérale de fer expose au risque de réactions anaphylactiques. Deux formes galéniques coexistent dont les modalités d’injection sont différentes et qu’il convient de bien distinguer lors de la prescription.
Anémie chez l’adulte et l’enfant
L’anémie de l’enfant est une question transversale récurrente. Les nombreuses étiologies permettent d’en faire une question insérée dans beaucoup de dossiers transversaux. L’axe principal sera le bilan diagnostique et la hiérarchisation des examens complémentaires en fonction des caractéristiques initiales de l’anémie. Par exemple, une anémie ferriprive peut faire diagnostiquer une carence d’apport à la fois par maltraitance mais aussi une malabsorption comme dans une maladie cœliaque, par exemple. Une anémie régénérative peut amener à diagnostiquer une hémolyse à anticorps froids dans le cadre d’une infection pulmonaire à mycoplasme. Par ailleurs, un dossier d’anémie peut être l’occasion de poser des questions sur les modalités et possibles complications des transfusions, notamment dans un dossier sur les hémoglobinopathies. Enfin, il est indispensable de connaître les modalités des traitements en cas de carence martiale, en B12 ou en folates. L’anémie de l’enfant est une question transversale récurrente. Les nombreuses étiologies permettent d’en faire une question insérée dans beaucoup de dossiers transversaux. L’axe principal sera le bilan diagnostique et la hiérarchisation des examens complémentaires en fonction des caractéristiques initiales de l’anémie. Par exemple, une anémie ferriprive peut faire diagnostiquer une carence d’apport à la fois par maltraitance mais aussi une malabsorption comme dans une maladie cœliaque, par exemple. Une anémie régénérative peut amener à diagnostiquer une hémolyse à anticorps froids dans le cadre d’une infection pulmonaire à mycoplasme. Par ailleurs, un dossier d’anémie peut être l’occasion de poser des questions sur les modalités et possibles complications des transfusions, notamment dans un dossier sur les hémoglobinopathies. Enfin, il est indispensable de connaître les modalités des traitements en cas de carence martiale, en B12 ou en folates.
Société française d’hématologie, référentiel. Abrégé d’hématologie, Elsevier Masson Ed.
The Anemias (chapitres 439 et suivants). Behrman, Kliegman, Jenson in Nelson Texbook of Pediatrics. Saunders Ed.
Haute Autorité de santé (HAS). Carence martiale. https://www.has-sante.fr/upload/docs/application/pdf/2011-11/texte_court__bilan_martial_carence_2011-11-09_17-22-2_135.pdf
HAS. Drépanocytose. https://www.has-sante.fr/upload/docs/application/pdf/2010-04/ald_10_pnds_drepano_enfant_web.pdf
HAS. Thalassémie. https://www.has-sante.fr/jcms/c_680242/fr/ald-n-10-syndromes-thalassemiques-majeurs-et-intermediaires
Société française de pédiatrie. Pondaré C. https://pap-pediatrie.fr/hematologie/anemies-du-jeune-enfant
Collège national des pédiatres universitaires (CNPU). http://campus.cerimes.fr/media/campus/deploiement/pediatrie/enseignement/anemie_fer_enfant/site/html/1.html
 Encadrés
Encadrés