La découverte des biothérapies, et plus récemment des petites molécules, a révolutionné la prise en charge et le pronostic d’enfants atteints d’arthrite juvénile idiopathique (AJI). Tandis que les biothérapies sont fabriquées par des biotechnologies et ciblent des cytokines ou des cellules impliquées dans la pathologie, les petites molécules sont des composés organiques qui inhibent un récepteur ou une enzyme.
Depuis la publication du premier essai randomisé contrôlé de l’anti-tumor necrosis factor (TNF) alpha étanercept dans l’AJI en 2000, plusieurs anticorps monoclonaux et protéines recombinantes humaines chimériques ont été développés et utilisés avec succès.
Les AJI constituent un groupe hétérogène de pathologies qui se définit selon l’International League of Associations for Rheumatology (ILAR) par la présence d’une arthrite de cause inconnue avant l’âge de 16 ans durant au moins six semaines et classifiée en sept sous-groupes différents (tableau 1 ).1
Afin de définir des sous-groupes plus homogènes, cette classification a récemment été révisée et inclut la forme systémique d’AJI (FS-AJI, ou maladie de Still), l’AJI avec facteurs antinucléaires (FAN) à début précoce (oligo- ou polyarticulaire), l’AJI avec facteur rhumatoïde, l’AJI avec enthésite, les autres AJI (ne correspondant à aucun des critères pour les sous-groupes décrits) et l’AJI non classée (plus d’un des critères) [tableau 1 ].2
Depuis la publication du premier essai randomisé contrôlé de l’anti-tumor necrosis factor (TNF) alpha étanercept dans l’AJI en 2000, plusieurs anticorps monoclonaux et protéines recombinantes humaines chimériques ont été développés et utilisés avec succès.
Les AJI constituent un groupe hétérogène de pathologies qui se définit selon l’International League of Associations for Rheumatology (ILAR) par la présence d’une arthrite de cause inconnue avant l’âge de 16 ans durant au moins six semaines et classifiée en sept sous-groupes différents (
Afin de définir des sous-groupes plus homogènes, cette classification a récemment été révisée et inclut la forme systémique d’AJI (FS-AJI, ou maladie de Still), l’AJI avec facteurs antinucléaires (FAN) à début précoce (oligo- ou polyarticulaire), l’AJI avec facteur rhumatoïde, l’AJI avec enthésite, les autres AJI (ne correspondant à aucun des critères pour les sous-groupes décrits) et l’AJI non classée (plus d’un des critères) [
Choix thérapeutique selon le type d’AJI
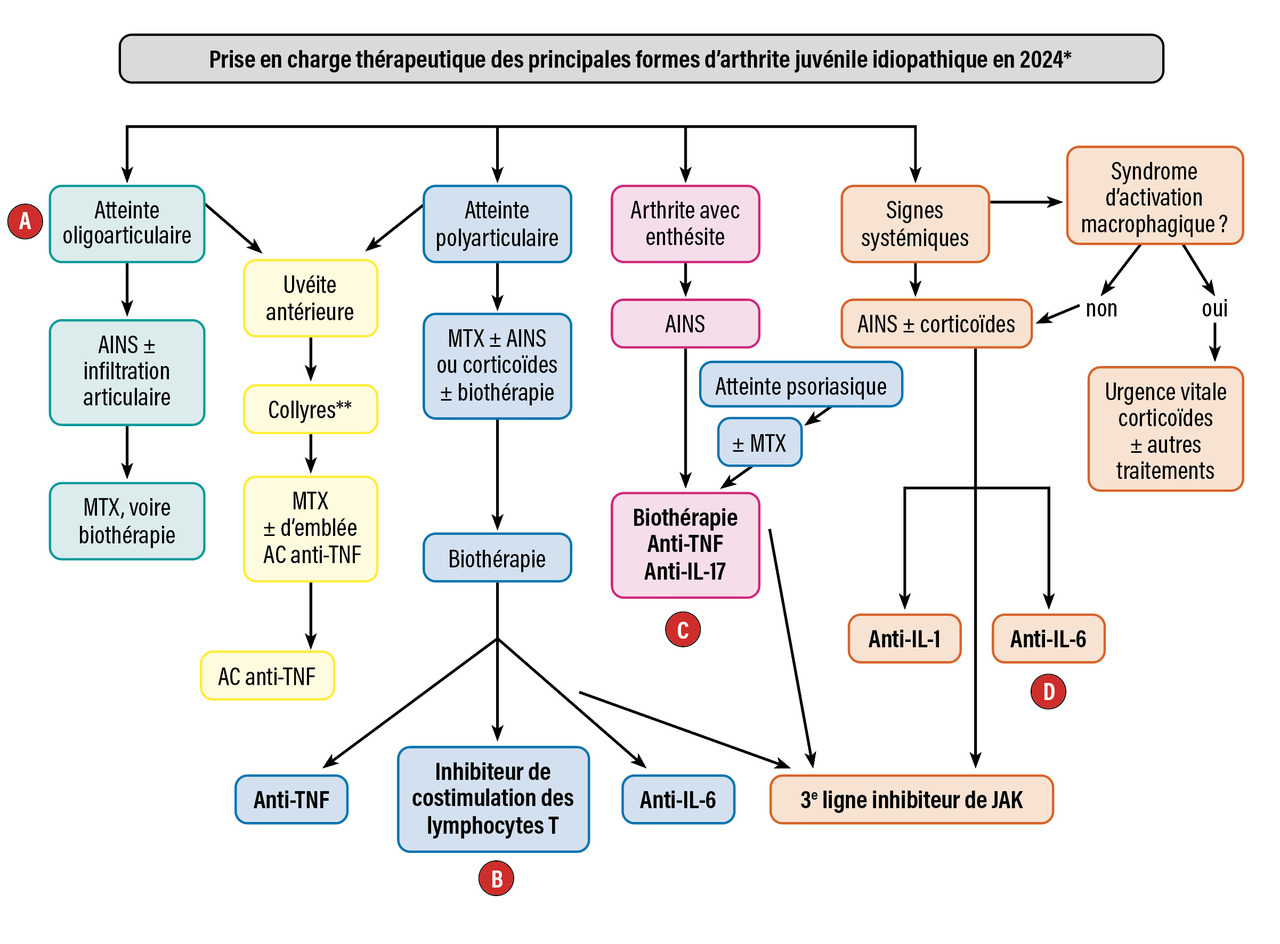
Le choix de la biothérapie et de la petite molécule dépend principalement du sous-groupe de l’AJI et des symptômes prédominants (arthrite, uvéite, signes systémiques) [figure ]. L’approche thérapeutique classique est d’utiliser les traitements conventionnels tels que les disease modifying antirheumatic drugs (DMARD ; par exemple, le méthotrexate [MTX]) en première intention et de considérer les biothérapies et petites molécules uniquement chez les patients non répondeurs. Mais, même si la plupart des autorisations de mise sur le marché (AMM) suivent encore le schéma classique, l’utilisation précoce des biothérapies est de plus en plus fréquente.
Les approches thérapeutiques actuelles favorisent la stratégie « treat-to-target » qui vise à obtenir rapidement une maladie inactive par initiation précoce de traitement immunosuppresseur incluant les biothérapies et petites molécules, avec une réévaluation régulière. Les différentes biothérapies et petites molécules autorisées ou à l’étude sont présentées en fonction des sous-groupes d’AJI dans letableau 2 .
Les approches thérapeutiques actuelles favorisent la stratégie « treat-to-target » qui vise à obtenir rapidement une maladie inactive par initiation précoce de traitement immunosuppresseur incluant les biothérapies et petites molécules, avec une réévaluation régulière. Les différentes biothérapies et petites molécules autorisées ou à l’étude sont présentées en fonction des sous-groupes d’AJI dans le
Lire aussi | Notre dossier – Arthrites juvéniles idiopathiques
Forme systémique d’AJI (maladie de Still)
La FS-AJI à début pédiatrique est caractérisée en début de maladie par la présence de signes généraux importants tels que la fièvre, l’éruption cutanée et la sérosite (lire article « Forme systémique d’arthrite juvénile idiopathique. Maladie de Still à début pédiatrique » page 1076 ).
Afin de permettre un diagnostic et une prise en charge thérapeutique plus précoces, l’arthrite n’est pas indispensable au diagnostic chez l’enfant selon les nouveaux critères de classification. Les complications, notamment la survenue d’un syndrome d’activation macrophagique et l’hypertension artérielle pulmonaire, peuvent mettre en jeu le pronostic vital.
La dérégulation de la réponse immunitaire innée avec surproduction de cytokines majeures telles que l’interleukine (IL)-1 et l’IL-6 joue un rôle prédominant dans la physiopathologie de la FS-JIA. En conséquence, les principaux progrès thérapeutiques récents sont liés à l’utilisation d’antagonistes de l’IL-1 (anakinra, canakinumab) et de l’IL-6 (tocilizumab, sarilumab [étude conduite mais pas encore publiée]).3,4 L’efficacité des anti-IL-1 et anti-IL-6 dans la FS-AJI est remarquable, une autorisation de mise sur le marché (AMM) a été obtenue pour l’anakinra, le canakinumab et le tocilizumab (formes intraveineuse [IV] dès l’âge de 2 ans et sous-cutanée [SC] dès l’âge de 1 an) dans cette indication.
Au vu des connaissances croissantes, les petites molécules, inhibiteurs de Janus kinase (JAK), baricitinib et tofacitinib, sont à l’essai dans la FS-AJI et l’anticorps anti-interféron gamma émapalumab dans le syndrome d’activation macrophagique sur FS-AJI, avec des résultats encourageants mais une efficacité qui reste à confirmer. Il existe un sous-groupe de patients caractérisé par un tableau hyperinflammatoire avec une IL-18 très élevée, des syndromes d’activation macrophagique récurrents et un risque augmenté d’atteinte pulmonaire qui peut justifier des approches thérapeutiques novatrices, en cours de discussion.5,6
Certains experts favorisent une introduction de l’anti-IL-1 anakinra beaucoup plus précoce, dans le but d’une rapide amélioration et d’une rémission complète sans corticostéroïdes après trois à six mois. Effectivement, des taux de réponse très élevés et des rémissions persistantes à l’arrêt de l’anakinra ont été rapportés chez des patients avec FS-AJI sans réponse aux anti-inflammatoires non stéroïdiens (AINS) après une introduction précoce d’anakinra, avant toute corticothérapie systémique.7 Ces observations sont en faveur d’une fenêtre d’opportunité (« window of opportunity ») au début de la maladie, pendant laquelle les patients sont potentiellement plus susceptibles au traitement ciblé tel que le blocage d’IL-1.
Afin de permettre un diagnostic et une prise en charge thérapeutique plus précoces, l’arthrite n’est pas indispensable au diagnostic chez l’enfant selon les nouveaux critères de classification. Les complications, notamment la survenue d’un syndrome d’activation macrophagique et l’hypertension artérielle pulmonaire, peuvent mettre en jeu le pronostic vital.
La dérégulation de la réponse immunitaire innée avec surproduction de cytokines majeures telles que l’interleukine (IL)-1 et l’IL-6 joue un rôle prédominant dans la physiopathologie de la FS-JIA. En conséquence, les principaux progrès thérapeutiques récents sont liés à l’utilisation d’antagonistes de l’IL-1 (anakinra, canakinumab) et de l’IL-6 (tocilizumab, sarilumab [étude conduite mais pas encore publiée]).3,4 L’efficacité des anti-IL-1 et anti-IL-6 dans la FS-AJI est remarquable, une autorisation de mise sur le marché (AMM) a été obtenue pour l’anakinra, le canakinumab et le tocilizumab (formes intraveineuse [IV] dès l’âge de 2 ans et sous-cutanée [SC] dès l’âge de 1 an) dans cette indication.
Au vu des connaissances croissantes, les petites molécules, inhibiteurs de Janus kinase (JAK), baricitinib et tofacitinib, sont à l’essai dans la FS-AJI et l’anticorps anti-interféron gamma émapalumab dans le syndrome d’activation macrophagique sur FS-AJI, avec des résultats encourageants mais une efficacité qui reste à confirmer. Il existe un sous-groupe de patients caractérisé par un tableau hyperinflammatoire avec une IL-18 très élevée, des syndromes d’activation macrophagique récurrents et un risque augmenté d’atteinte pulmonaire qui peut justifier des approches thérapeutiques novatrices, en cours de discussion.5,6
Certains experts favorisent une introduction de l’anti-IL-1 anakinra beaucoup plus précoce, dans le but d’une rapide amélioration et d’une rémission complète sans corticostéroïdes après trois à six mois. Effectivement, des taux de réponse très élevés et des rémissions persistantes à l’arrêt de l’anakinra ont été rapportés chez des patients avec FS-AJI sans réponse aux anti-inflammatoires non stéroïdiens (AINS) après une introduction précoce d’anakinra, avant toute corticothérapie systémique.7 Ces observations sont en faveur d’une fenêtre d’opportunité (« window of opportunity ») au début de la maladie, pendant laquelle les patients sont potentiellement plus susceptibles au traitement ciblé tel que le blocage d’IL-1.
AJI à début non systémique
Il s’agit des AJI avec facteurs antinucléaires (FAN) à début précoce (oligo- ou polyarticulaire), des AJI avec facteur rhumatoïde, des AJI avec enthésite, des autres AJI et des AJI non classées.
AJI avec FAN à début précoce (oligo- ou polyarticulaire)
Ces patients ont un risque d’uvéite antérieure chronique asymptomatique (ou « uvéite à œil blanc »). Cela impose un examen ophtalmologique à la lampe à fente systématique tous les trois mois pendant les cinq premières années après le diagnostic afin de permettre une détection précoce de l’atteinte oculaire et d’éviter des complications irréversibles (synéchies, cataracte, glaucome, diminution de la vision jusqu’à la cécité).
Historiquement, les essais cliniques évaluant l’efficacité et la tolérance des biothérapies ont inclus principalement des patients avec AJI et atteinte polyarticulaire, donc un groupe hétérogène de patients tels que ceux atteints d’AJI à début oligoarticulaire secondairement étendu, d’AJI à début polyarticulaire avec ou sans facteur rhumatoïde, d’arthrite juvénile liée à l’enthésite avec atteinte périphérique et d’AJI liée au psoriasis. Cette entité fonctionnelle d’AJI avec atteinte polyarticulaire est présentée ci-dessous.
L’infliximab n’ayant pas atteint l’objectif principal de l’essai clinique pour raisons méthodologiques, il n’a pas d’AMM, bien que son efficacité semble similaire à celle de l’étanercept et de l’adalimumab.
Le golimumab a une AMM en association avec le MTX, pour les AJI avec atteinte polyarticulaire qui n’ont pas répondu de manière adéquate à un précédent traitement par MTX.11
Dans une étude de phase II, le tocilizumab sous-cutané n’a pas permis de diminuer l’inflammation oculaire significativement chez des patients réfractaires au MTX et/ou à l’adalimumab. Néanmoins, il semble avoir un intérêt chez certains patients difficiles à traiter, notamment sur l’œdème maculaire.18
Un essai clinique avec l’inhibiteur de JAK baricitinib n’a pas démontré son efficacité chez un petit groupe de patients réfractaires (communication au congrès ACR 2024).
Historiquement, les essais cliniques évaluant l’efficacité et la tolérance des biothérapies ont inclus principalement des patients avec AJI et atteinte polyarticulaire, donc un groupe hétérogène de patients tels que ceux atteints d’AJI à début oligoarticulaire secondairement étendu, d’AJI à début polyarticulaire avec ou sans facteur rhumatoïde, d’arthrite juvénile liée à l’enthésite avec atteinte périphérique et d’AJI liée au psoriasis. Cette entité fonctionnelle d’AJI avec atteinte polyarticulaire est présentée ci-dessous.
Anti-TNF alpha
Les anti-TNF alpha ont été les premières biothérapies à obtenir une AMM dans l’AJI au début des années 2000. L’étanercept, récepteur soluble du TNF-alpha, puis l’infliximab et l’adalimumab, des anticorps monoclonaux, ont démontré leur efficacité dans les AJI avec atteinte polyarticulaire réfractaire ou intolérante au MTX.8-10 Une AMM a été obtenue pour l’étanercept et l’adalimumab dans ces indications. Des effets positifs ont été démontrés également sur la croissance, la qualité de vie et la réponse à long terme.L’infliximab n’ayant pas atteint l’objectif principal de l’essai clinique pour raisons méthodologiques, il n’a pas d’AMM, bien que son efficacité semble similaire à celle de l’étanercept et de l’adalimumab.
Le golimumab a une AMM en association avec le MTX, pour les AJI avec atteinte polyarticulaire qui n’ont pas répondu de manière adéquate à un précédent traitement par MTX.11
Abatacept
L’abatacept, inhibiteur de la costimulation qui cible CTLA-4 et inactive les lymphocytes T, a démontré son efficacité chez des patients avec AJI et atteinte polyarticulaire en échec ou intolérance au MTX ou à un autre DMARD incluant les anti-TNF.12 Une AMM a été obtenue pour les formes IV (dès l’âge de 6 ans) et SC (dès l’âge de 2 ans) dans cette indication. Des effets positifs ont aussi été démontrés sur l’efficacité et la qualité de vie à long terme.Anti-IL-6
Le tocilizumab, un anticorps anti-récepteur de l’IL-6, a démontré son efficacité dans les AJI avec atteinte polyarticulaire en échec de MTX ou d’une première biothérapie.13 Une AMM pour les formes IV (dès l’âge de 2 ans) et SC (dès l’âge de 1 an) existe dans cette indication. Un autre anticorps monoclonal antirécepteur de l’IL-6, le sarilumab, a été étudié par voie SC (les résultats n’ont pas encore été publiés).Inhibiteurs de JAK
À la suite des résultats encourageants d’essais cliniques, le tofacitinib, un inhibiteur de JAK 1/3, et le baricitinib, un inhibiteur de JAK 1/3, ont obtenu une AMM pour les AJI avec atteinte polyarticulaire en échec d’un DMARD.14,15 Néanmoins, il est proposé de ne les utiliser qu’en troisième ligne après au moins deux traitements de fond conventionnels ou biologiques en raison de potentiels effets indésirables (risque de thrombose, risque oncologique chez l’adulte fumeur). L’avantage des inhibiteurs de JAK est la prise par voie orale ; néanmoins, l’expérience dans les AJI est encore limitée et récente, et des données de pharmacovigilance et de tolérance à long terme sont nécessaires.Uvéites chroniques antérieures associées à l’AJI
Contrairement à l’étanercept, les anticorps monoclonaux ont un intérêt dans le traitement des uvéites chroniques antérieures associées à l’AJI. En particulier, l’adalimumab en association au MTX a démontré une efficacité très significative permettant l’obtention d’une AMM dans cette indication.16,17Dans une étude de phase II, le tocilizumab sous-cutané n’a pas permis de diminuer l’inflammation oculaire significativement chez des patients réfractaires au MTX et/ou à l’adalimumab. Néanmoins, il semble avoir un intérêt chez certains patients difficiles à traiter, notamment sur l’œdème maculaire.18
Un essai clinique avec l’inhibiteur de JAK baricitinib n’a pas démontré son efficacité chez un petit groupe de patients réfractaires (communication au congrès ACR 2024).
AJI avec facteur rhumatoïde
L’AJI avec facteur rhumatoïde représente la forme juvénile de la polyarthrite rhumatoïde de l’adulte. Il n’existe aucun essai clinique évaluant spécifiquement l’AJI avec facteur rhumatoïde. Le traitement est identique aux AJI avec atteinte polyarticulaire ; au cas par cas, l’anticorps anti-CD20 rituximab est discuté dans les AJI avec facteur rhumatoïde ; il n’existe pas d’AMM dans cette indication.
AJI avec enthésite
La présentation de l’AJI avec enthésite, un sous-groupe ressemblant aux spondylarthrites de l’adulte, est caractérisée par une arthrite périphérique avec ou sans enthésite. L’inflammation de la colonne vertébrale peut survenir secondairement. Les anti-TNF étanercept et adalimumab ont démontré un effet positif sur l’atteinte périphérique et axiale ; ils ont une AMM chez ces patients en cas de réponse insuffisante ou d’intolérance au traitement conventionnel.19,20 Récemment, le sécukinumab, un anticorps anti-IL-17A, a obtenu une AMM dans cette même indication chez des enfants dès l’âge de 6 ans21 et le baricitinib pour les AJI avec enthésite avec réponse inadéquate ou intolérance à au moins un DMARD conventionnel ou biologique antérieur.15
Surveiller la tolérance des biothérapies et des petites molécules
La tolérance des biothérapies est globalement satisfaisante sur le moyen terme ; mais des effets indésirables infectieux, dysimmunitaires (par exemple lésions psoriasiformes, atteintes neurologiques démyélinisantes, maladies intestinales inflammatoires, apparition d’auto-anticorps sans relevance clinique) et allergiques ont été observés.
Le type de la biothérapie, les immunosuppresseurs concomitants (en particulier les corticostéroïdes), la pathologie sous-jacente et l’activité de la maladie semblent influencer le risque d’infection. Il s’agit en particulier des herpès virus et de la tuberculose mais aussi d’infections bactériennes qui peuvent être sévères et potentiellement fatales.22
Les vaccins jouent un rôle primordial dans la protection vis-à-vis de ces risques infectieux, notamment la vaccination antivaricelle en cas d’absence d’immunité, la vaccination antipneumococcique, antiméningococcique et antigrippale annuelle. Les vaccins vivants sont théoriquement contre-indiqués sous traitement immunosuppresseur. Mais, contrairement aux patients avec déficit immunitaire primitif, les vaccins vivants atténués comme celui de la varicelle ou le ROR (rougeole, oreillons, rubéole) ne sont pas associés à des infections vaccinales graves chez des patients sous traitement de fond par DMARD ou biothérapie – sauf en cas de forte corticothérapie systémique associée –, raison pour laquelle ils sont discutés au cas par cas en évaluant le rapport bénéfice-risque.23
Des réactions d’intolérance très particulières aux biothérapies pouvant mimer dans certains cas un DRESS (drug reaction with eosinophilia and systemic symptoms) ont été rapportés chez des patients avec FS-AJI et atteinte pulmonaire.
Enfin, le potentiel risque accru de néoplasie, notamment de lymphome, a longuement été discuté. Les patients avec AJI semblent avoir un risque deux à quatre fois plus élevé de développer un cancer par rapport à la population générale, mais ce risque ne semble pas être associé aux biothérapies. Chez l’adulte, un excès de cancers cutanés, en particulier de carcinomes basocellulaires, a été rapporté.
Les effets indésirables principaux des inhibiteurs de JAK incluent les infections potentiellement sévères (dont le zona), les cytopénies, l’intolérance hépatique, les thromboses et de possibles néoplasies.
Différents registres de patients avec AJI sont mis en place par les laboratoires pharmaceutiques et par les rhumatologues pédiatres afin de permettre un suivi de l’efficacité et une pharmacovigilance sur le long terme pour mieux connaître les risques associés aux biothérapies.
L’utilisation des biothérapies et des petites molécules nécessite une bonne connaissance des différents agents disponibles, le choix est guidé par la pathologie de l’enfant (AMM), le mode d’administration (SC versus IV, oral pour les inhibiteurs de JAK) et les possibles effets indésirables. Il n’existe pas d’étude comparative des biothérapies dans l’AJI. La durée du traitement est dépendante du sous-type de l’AJI (la plus courte possible pour les FS-AJI en cas de début très précoce de l’anti-IL-1, prolongée en cas d’uvéite) et de l’évolution (traitement prolongé pour les formes sévères en échec de multiples DMARD).
Le type de la biothérapie, les immunosuppresseurs concomitants (en particulier les corticostéroïdes), la pathologie sous-jacente et l’activité de la maladie semblent influencer le risque d’infection. Il s’agit en particulier des herpès virus et de la tuberculose mais aussi d’infections bactériennes qui peuvent être sévères et potentiellement fatales.22
Les vaccins jouent un rôle primordial dans la protection vis-à-vis de ces risques infectieux, notamment la vaccination antivaricelle en cas d’absence d’immunité, la vaccination antipneumococcique, antiméningococcique et antigrippale annuelle. Les vaccins vivants sont théoriquement contre-indiqués sous traitement immunosuppresseur. Mais, contrairement aux patients avec déficit immunitaire primitif, les vaccins vivants atténués comme celui de la varicelle ou le ROR (rougeole, oreillons, rubéole) ne sont pas associés à des infections vaccinales graves chez des patients sous traitement de fond par DMARD ou biothérapie – sauf en cas de forte corticothérapie systémique associée –, raison pour laquelle ils sont discutés au cas par cas en évaluant le rapport bénéfice-risque.23
Des réactions d’intolérance très particulières aux biothérapies pouvant mimer dans certains cas un DRESS (drug reaction with eosinophilia and systemic symptoms) ont été rapportés chez des patients avec FS-AJI et atteinte pulmonaire.
Enfin, le potentiel risque accru de néoplasie, notamment de lymphome, a longuement été discuté. Les patients avec AJI semblent avoir un risque deux à quatre fois plus élevé de développer un cancer par rapport à la population générale, mais ce risque ne semble pas être associé aux biothérapies. Chez l’adulte, un excès de cancers cutanés, en particulier de carcinomes basocellulaires, a été rapporté.
Les effets indésirables principaux des inhibiteurs de JAK incluent les infections potentiellement sévères (dont le zona), les cytopénies, l’intolérance hépatique, les thromboses et de possibles néoplasies.
Différents registres de patients avec AJI sont mis en place par les laboratoires pharmaceutiques et par les rhumatologues pédiatres afin de permettre un suivi de l’efficacité et une pharmacovigilance sur le long terme pour mieux connaître les risques associés aux biothérapies.
L’utilisation des biothérapies et des petites molécules nécessite une bonne connaissance des différents agents disponibles, le choix est guidé par la pathologie de l’enfant (AMM), le mode d’administration (SC versus IV, oral pour les inhibiteurs de JAK) et les possibles effets indésirables. Il n’existe pas d’étude comparative des biothérapies dans l’AJI. La durée du traitement est dépendante du sous-type de l’AJI (la plus courte possible pour les FS-AJI en cas de début très précoce de l’anti-IL-1, prolongée en cas d’uvéite) et de l’évolution (traitement prolongé pour les formes sévères en échec de multiples DMARD).
Des limites à ces nouveaux traitements persistent
Les biothérapies et petites molécules ont révolutionné la prise en charge et le pronostic d’enfants avec AJI et jouent un rôle de plus en plus important dans les différentes formes d’AJI. La prise en charge en centre de référence et de compétences pédiatriques (www.fai2r.org) en lien avec le pédiatre ou le médecin traitant et les services de santé de proximité est important. Néanmoins, certaines difficultés persistent : le manque de biomarqueurs, en cours de recherche, qui aident à choisir la biothérapie ou la petite molécule la plus efficace pour un certain patient et à définir quand arrêter le traitement une fois que le patient est en rémission (médecine personnalisée), la nécessité d’une meilleure connaissance de la tolérance à long terme et la mise à disposition de traitements efficaces pour les patients qui ne répondent pas adéquatement aux agents thérapeutiques actuellement disponibles.
Références
1. Petty RE, Southwood TR, Manners P, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: Second revision, Edmonton, 2001. J Rheumatol. 2004;31:390-2.
2. Martini A, Ravelli A, Avcin T, , et al. Toward new classification criteria for juvenile idiopathic arthritis: First steps, Pediatric Rheumatology International Trials Organization International Consensus. J Rheumatol 2019;46:190-7.
3. Ruperto N, Brunner HI, Quartier P, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med 2012;367:2396-406.
4. De Benedetti F, Brunner HI, Ruperto N, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367:2385-95.
5. Saper VE, Chen G, Deutsch GH, et al. Emergent high fatality lung disease in systemic juvenile arthritis. Ann Rheum Dis 2019;78:1722-31.
6. Fautrel B, Mitrovic S, De Matteis A, et al. EULAR/PReS recommendations for the diagnosis and management of Still’s disease, comprising systemic juvenile idiopathic arthritis and adult-onset Still’s disease. Ann Rheum Dis 2024;83:1614-27.
7. Vastert SJ, de Jager W, Noordman BJ, et al. Effectiveness of first-line treatment with recombinant interleukin-1 receptor antagonist in steroid-naive patients with new-onset systemic juvenile idiopathic arthritis: Results of a prospective cohort study. Arthritis Rheumatol 2014;66:1034-43.
8. Lovell DJ, Giannini EH, Reiff A, et al. Etanercept in children with polyarticular juvenile rheumatoid arthritis. Pediatric Rheumatology Collaborative Study Group. N Engl J Med 2000;342:763-9.
9. Lovell DJ, Ruperto N, Goodman S, et al. Adalimumab with or without methotrexate in juvenile rheumatoid arthritis. N Engl J Med 2008;359:810-20.
10. Ruperto N, Lovell DJ, Cuttica R, et al. A randomized, placebo-controlled trial of infliximab plus methotrexate for the treatment of polyarticular-course juvenile rheumatoid arthritis. Arthritis Rheum 2007;56:3096-106.
11. Brunner HI, Ruperto N, Tzaribachev N, et al. Subcutaneous golimumab for children with active polyarticular-course juvenile idiopathic arthritis: Results of a multicentre, double-blind, randomised-withdrawal trial. Ann Rheum Dis 2018;77:21-9.
12. Ruperto N, Lovell DJ, Quartier P, et al. Abatacept in children with juvenile idiopathic arthritis: A randomised, double-blind, placebo-controlled withdrawal trial. Lancet 2008;372:383-91.
13. Brunner HI, Ruperto N, Zuber Z, et al. Efficacy and safety of tocilizumab in patients with polyarticular-course juvenile idiopathic arthritis: Results from a phase 3, randomised, double-blind withdrawal trial. Ann Rheum Dis 2015;74:1110-7.
14. Ruperto N, Brunner HI, Synoverska O, et al. Tofacitinib in juvenile idiopathic arthritis: A double-blind, placebo-controlled, withdrawal phase 3 randomised trial. Lancet 2021;398:1984-96.
15. Ramanan AV, Quartier P, Okamoto N, et al. Baricitinib in juvenile idiopathic arthritis: An international, phase 3, randomised, double-blind, placebo-controlled, withdrawal, efficacy, and safety trial. Lancet 2023;402:555-70.
16. Ramanan AV, Dick AD, Beresford MW. Adalimumab for uveitis in juvenile idiopathic arthritis. N Engl J Med 2017;377:789-90.
17. Quartier P, Baptiste A, Despert V, et al. ADJUVITE: A double-blind, randomised, placebo-controlled trial of adalimumab in early onset, chronic, juvenile idiopathic arthritis-associated anterior uveitis. Ann Rheum Dis 2018;77:1003-11.
18. Ramanan AV, Dick AD, Guly C, et al. Tocilizumab in patients with anti-TNF refractory juvenile idiopathic arthritis-associated uveitis (APTITUDE): A multicentre, single-arm, phase 2 trial. Lancet Rheumatol 2020;2:e135-e41.
19. Horneff G, Fitter S, Foeldvari I, et al. Double-blind, placebo-controlled randomized trial with adalimumab for treatment of juvenile onset ankylosing spondylitis (JoAS): Significant short term improvement. Arthritis Res Ther 2012;14:R230.
20. Horneff G, Foeldvari I, Minden K, et al. Efficacy and safety of etanercept in patients with the enthesitis-related arthritis category of juvenile idiopathic arthritis: Results from a phase III randomized, double-blind study. Arthritis Rheumatol 2015;67:2240-9.
21. Brunner HI, Foeldvari I, Alexeeva E, et al. Secukinumab in enthesitis-related arthritis and juvenile psoriatic arthritis: A randomised, double-blind, placebo-controlled, treatment withdrawal, phase 3 trial. Ann Rheum Dis 2023;82:154-60.
22. Aeschlimann FA, Chong SL, Lyons TW, et al. Risk of serious infections associated with biologic agents in juvenile idiopathic arthritis: A systematic review and meta-analyses. J Pediatr 2019;204:162-71.e3.
23. Jansen MHA, Rondaan C, Legger GE, et al. EULAR/PRES recommendations for vaccination of paediatric patients with autoimmune inflammatory rheumatic diseases: Update 2021. Ann Rheum Dis 2023;82:35-47.
2. Martini A, Ravelli A, Avcin T, , et al. Toward new classification criteria for juvenile idiopathic arthritis: First steps, Pediatric Rheumatology International Trials Organization International Consensus. J Rheumatol 2019;46:190-7.
3. Ruperto N, Brunner HI, Quartier P, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med 2012;367:2396-406.
4. De Benedetti F, Brunner HI, Ruperto N, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367:2385-95.
5. Saper VE, Chen G, Deutsch GH, et al. Emergent high fatality lung disease in systemic juvenile arthritis. Ann Rheum Dis 2019;78:1722-31.
6. Fautrel B, Mitrovic S, De Matteis A, et al. EULAR/PReS recommendations for the diagnosis and management of Still’s disease, comprising systemic juvenile idiopathic arthritis and adult-onset Still’s disease. Ann Rheum Dis 2024;83:1614-27.
7. Vastert SJ, de Jager W, Noordman BJ, et al. Effectiveness of first-line treatment with recombinant interleukin-1 receptor antagonist in steroid-naive patients with new-onset systemic juvenile idiopathic arthritis: Results of a prospective cohort study. Arthritis Rheumatol 2014;66:1034-43.
8. Lovell DJ, Giannini EH, Reiff A, et al. Etanercept in children with polyarticular juvenile rheumatoid arthritis. Pediatric Rheumatology Collaborative Study Group. N Engl J Med 2000;342:763-9.
9. Lovell DJ, Ruperto N, Goodman S, et al. Adalimumab with or without methotrexate in juvenile rheumatoid arthritis. N Engl J Med 2008;359:810-20.
10. Ruperto N, Lovell DJ, Cuttica R, et al. A randomized, placebo-controlled trial of infliximab plus methotrexate for the treatment of polyarticular-course juvenile rheumatoid arthritis. Arthritis Rheum 2007;56:3096-106.
11. Brunner HI, Ruperto N, Tzaribachev N, et al. Subcutaneous golimumab for children with active polyarticular-course juvenile idiopathic arthritis: Results of a multicentre, double-blind, randomised-withdrawal trial. Ann Rheum Dis 2018;77:21-9.
12. Ruperto N, Lovell DJ, Quartier P, et al. Abatacept in children with juvenile idiopathic arthritis: A randomised, double-blind, placebo-controlled withdrawal trial. Lancet 2008;372:383-91.
13. Brunner HI, Ruperto N, Zuber Z, et al. Efficacy and safety of tocilizumab in patients with polyarticular-course juvenile idiopathic arthritis: Results from a phase 3, randomised, double-blind withdrawal trial. Ann Rheum Dis 2015;74:1110-7.
14. Ruperto N, Brunner HI, Synoverska O, et al. Tofacitinib in juvenile idiopathic arthritis: A double-blind, placebo-controlled, withdrawal phase 3 randomised trial. Lancet 2021;398:1984-96.
15. Ramanan AV, Quartier P, Okamoto N, et al. Baricitinib in juvenile idiopathic arthritis: An international, phase 3, randomised, double-blind, placebo-controlled, withdrawal, efficacy, and safety trial. Lancet 2023;402:555-70.
16. Ramanan AV, Dick AD, Beresford MW. Adalimumab for uveitis in juvenile idiopathic arthritis. N Engl J Med 2017;377:789-90.
17. Quartier P, Baptiste A, Despert V, et al. ADJUVITE: A double-blind, randomised, placebo-controlled trial of adalimumab in early onset, chronic, juvenile idiopathic arthritis-associated anterior uveitis. Ann Rheum Dis 2018;77:1003-11.
18. Ramanan AV, Dick AD, Guly C, et al. Tocilizumab in patients with anti-TNF refractory juvenile idiopathic arthritis-associated uveitis (APTITUDE): A multicentre, single-arm, phase 2 trial. Lancet Rheumatol 2020;2:e135-e41.
19. Horneff G, Fitter S, Foeldvari I, et al. Double-blind, placebo-controlled randomized trial with adalimumab for treatment of juvenile onset ankylosing spondylitis (JoAS): Significant short term improvement. Arthritis Res Ther 2012;14:R230.
20. Horneff G, Foeldvari I, Minden K, et al. Efficacy and safety of etanercept in patients with the enthesitis-related arthritis category of juvenile idiopathic arthritis: Results from a phase III randomized, double-blind study. Arthritis Rheumatol 2015;67:2240-9.
21. Brunner HI, Foeldvari I, Alexeeva E, et al. Secukinumab in enthesitis-related arthritis and juvenile psoriatic arthritis: A randomised, double-blind, placebo-controlled, treatment withdrawal, phase 3 trial. Ann Rheum Dis 2023;82:154-60.
22. Aeschlimann FA, Chong SL, Lyons TW, et al. Risk of serious infections associated with biologic agents in juvenile idiopathic arthritis: A systematic review and meta-analyses. J Pediatr 2019;204:162-71.e3.
23. Jansen MHA, Rondaan C, Legger GE, et al. EULAR/PRES recommendations for vaccination of paediatric patients with autoimmune inflammatory rheumatic diseases: Update 2021. Ann Rheum Dis 2023;82:35-47.
Dans cet article

Résumé
L’introduction des biothérapies a révolutionné la prise en charge des enfants atteints d’arthrite juvénile idiopathique (AJI). Ces biothérapies, ciblant le tumor necrosis factor (TNF) alpha, l’interleukine (IL)-1, l’IL-6, l’IL-17 ou les signaux de costimulation lymphocytaires T (abatacept), et plus récemment les petites molécules, les inhibiteurs de Janus kinase (JAK), ont amélioré significativement le pronostic des patients. De plus, de nombreuses biothérapies sont actuellement à l’essai. La tolérance globale des agents biologiques est similaire à celle observée chez l’adulte ; les effets indésirables sont surtout de nature infectieuse, dysimmunitaire et allergique. La mise à jour des vaccinations est décisive dans la prévention des complications liées au traitement. La prise en charge des enfants atteints d’AJI sous biothérapie dans un centre spécialisé, le suivi de l’efficacité et la pharmacovigilance sur le long terme sont importants.