L’hypertension pulmonaire (HTP) associée aux maladies respiratoires chroniques est fréquente chez les patients présentant une bronchopneumopathie chronique obstructive (BPCO) avec ou sans emphysème, une pneumopathie interstitielle diffuse (PID), un syndrome emphysème-fibrose ou encore une hypoventilation alvéolaire. Ces HTP appartiennent au groupe 3 de la classification clinique (tableau 1 ). Leur diagnostic est établi sur les données d’un cathétérisme cardiaque droit réalisé dans un centre de compétence, chez un patient à l’état stable. Conformément aux nouvelles recommandations de prise en charge,1 le diagnostic d’HTP est retenu si la pression artérielle pulmonaire moyenne (PAPm) est supérieure à 20 mmHg, les résistances vasculaires pulmonaires (RVP) supérieures à 2 unités Wood (UW) et la pression artérielle pulmonaire d’occlusion (PAPO) inférieure ou égale à 15 mmHg. Cependant, le cathétérisme cardiaque droit n’est proposé que si l’on soupçonne que les résultats modifieront la prise en charge.
Lorsque les RVP sont supérieures à 5 UW, le pronostic à court terme est particulièrement sévère : parmi tous les patients avec HTP, les patients du groupe 3 sont ceux dont l’espérance de vie est la plus courte. Dans le registre ASPIRE (Sheffield, Royaume-Uni), ces patients avaient seulement 41 % de survie à trois ans.2 Ce mauvais pronostic justifie leur adressage à un centre de compétence ou de référence de l’HTP pour discuter de l’introduction d’un traitement spécifique.
Lorsque les RVP sont supérieures à 5 UW, le pronostic à court terme est particulièrement sévère : parmi tous les patients avec HTP, les patients du groupe 3 sont ceux dont l’espérance de vie est la plus courte. Dans le registre ASPIRE (Sheffield, Royaume-Uni), ces patients avaient seulement 41 % de survie à trois ans.2 Ce mauvais pronostic justifie leur adressage à un centre de compétence ou de référence de l’HTP pour discuter de l’introduction d’un traitement spécifique.
Physiopathologie mal élucidée
Les mécanismes physiopathologiques impliqués dans la survenue d’une HTP dans un contexte de maladie respiratoire chronique sont complexes et incomplètement élucidés. Il existe une raréfaction du lit vasculaire pulmonaire par les lésions d’emphysème ou de fibrose. L’hypoxémie chronique favorise la vasoconstriction hypoxique. De plus, un déséquilibre entre des facteurs pro- et antiangiogéniques, des anomalies moléculaires et génétiques concourent à la survenue d’un remodelage vasculaire pulmonaire.
Poser le diagnostic
Il est important de différencier une HTP légère à modérée (situation fréquente dans les maladies respiratoires chroniques évoluées) d’une HTP sévère. Cette dernière peut nécessiter une prise en charge particulière.
Certaines caractéristiques cliniques, fonctionnelles, biologiques et scanographiques permettent de suspecter une HTP.3-5 Cependant, il est toujours difficile de distinguer les signes cliniques en rapport avec l’affection causale de ceux de l’HTP. La dyspnée d’effort est en général sévère lorsqu’il y a une HTP compliquant une maladie respiratoire chronique. Il est également possible d’observer des signes d’insuffisance cardiaque droite (œdèmes des membres inférieurs, turgescences des veines jugulaires et reflux hépatojugulaire). Mais ces signes ne sont ni sensibles ni spécifiques.
Les explorations fonctionnelles respiratoires peuvent mettre en évidence un trouble sévère de la diffusion alvéolocapillaire (capacité de diffusion du monoxyde de carbone [DLCO], coefficient de transfert [KCO] ≤ 40 %, aggravation de la distance parcourue sur six minutes et aggravation de la désaturation).
Les examens d’imagerie (échocardiographie, scanner thoracique et imagerie par résonance magnétique [IRM] cardiaques) montrent une dilatation de l’artère pulmonaire et des cavités cardiaques droites.
De plus, le NT-proBNP peut être augmenté.
En pratique, c’est le plus souvent à l’occasion de la réalisation d’une échocardiographie demandée soit pour rechercher des signes d’hypertension pulmonaire, soit dans le cadre d’une comorbidité cardiaque gauche qu’une pression artérielle pulmonaire (PAP) systolique anormalement élevée est mise en évidence.
Avant de décider de réaliser un cathétérisme cardiaque droit, l’examen clinique et les examens paracliniques non invasifs peuvent suggérer la présence d’une HTP sévère. Une HTP peut être expliquée par une insuffisance respiratoire très sévère ou peut être le cas d’une BPCO associée à une obésité morbide et un trouble respiratoire du sommeil.
Le cathétérisme cardiaque droit n’est pas systématique en cas de suspicion d’HTP dans cette population. Il doit être réalisé si les résultats doivent faciliter les décisions de prise en charge. C’est le cas si un traitement chirurgical tel que la transplantation pulmonaire est envisagé, devant la suspicion de l’association d’une hypertension artérielle pulmonaire (HTAP) ou d’une HTP postembolique chronique. Cet examen est également recommandé lorsqu’un phénotype vasculaire pulmonaire est suspecté ou s’il est impossible de différencier une HTP sévère associée à une maladie respiratoire chronique d’une HTP associée à une cardiopathie gauche.
Certaines caractéristiques cliniques, fonctionnelles, biologiques et scanographiques permettent de suspecter une HTP.3-5 Cependant, il est toujours difficile de distinguer les signes cliniques en rapport avec l’affection causale de ceux de l’HTP. La dyspnée d’effort est en général sévère lorsqu’il y a une HTP compliquant une maladie respiratoire chronique. Il est également possible d’observer des signes d’insuffisance cardiaque droite (œdèmes des membres inférieurs, turgescences des veines jugulaires et reflux hépatojugulaire). Mais ces signes ne sont ni sensibles ni spécifiques.
Les explorations fonctionnelles respiratoires peuvent mettre en évidence un trouble sévère de la diffusion alvéolocapillaire (capacité de diffusion du monoxyde de carbone [DLCO], coefficient de transfert [KCO] ≤ 40 %, aggravation de la distance parcourue sur six minutes et aggravation de la désaturation).
Les examens d’imagerie (échocardiographie, scanner thoracique et imagerie par résonance magnétique [IRM] cardiaques) montrent une dilatation de l’artère pulmonaire et des cavités cardiaques droites.
De plus, le NT-proBNP peut être augmenté.
En pratique, c’est le plus souvent à l’occasion de la réalisation d’une échocardiographie demandée soit pour rechercher des signes d’hypertension pulmonaire, soit dans le cadre d’une comorbidité cardiaque gauche qu’une pression artérielle pulmonaire (PAP) systolique anormalement élevée est mise en évidence.
Avant de décider de réaliser un cathétérisme cardiaque droit, l’examen clinique et les examens paracliniques non invasifs peuvent suggérer la présence d’une HTP sévère. Une HTP peut être expliquée par une insuffisance respiratoire très sévère ou peut être le cas d’une BPCO associée à une obésité morbide et un trouble respiratoire du sommeil.
Le cathétérisme cardiaque droit n’est pas systématique en cas de suspicion d’HTP dans cette population. Il doit être réalisé si les résultats doivent faciliter les décisions de prise en charge. C’est le cas si un traitement chirurgical tel que la transplantation pulmonaire est envisagé, devant la suspicion de l’association d’une hypertension artérielle pulmonaire (HTAP) ou d’une HTP postembolique chronique. Cet examen est également recommandé lorsqu’un phénotype vasculaire pulmonaire est suspecté ou s’il est impossible de différencier une HTP sévère associée à une maladie respiratoire chronique d’une HTP associée à une cardiopathie gauche.
Lire aussi | Consensus sur l’hypertension pulmonaire
Approche thérapeutique globale
L’approche thérapeutique de l’HTP du groupe 3 commence par l’optimisation du traitement de la maladie pulmonaire sous-jacente et des comorbidités, la mise à jour des vaccinations (pneumocoque, grippe, SARS-CoV-2 et virus respiratoire syncytial [VRS]), la réadaptation à l’effort, voire le recours à la transplantation.
Il est notamment important de vérifier l’efficacité et l’observance d’une oxygénothérapie de longue durée et éventuellement d’une ventilation non invasive. Il existe des preuves limitées et contradictoires sur l’utilisation des médicaments approuvés pour le traitement de l’HTAP chez les patients atteints d’HTP du groupe 3.
Il est notamment important de vérifier l’efficacité et l’observance d’une oxygénothérapie de longue durée et éventuellement d’une ventilation non invasive. Il existe des preuves limitées et contradictoires sur l’utilisation des médicaments approuvés pour le traitement de l’HTAP chez les patients atteints d’HTP du groupe 3.
Particularités de l’HTP sur la BPCO (groupe 3.1)
La prévalence de l’HTP chez les patients présentant une BPCO est élevée puisqu’elle atteint près de 39 % des patients et 90 % des BPCO classés GOLD (Global initiative for chronic Obstructive Lung Disease) IV ou ayant un emphysème sévère ont une PAPm supérieure à 20 mmHg. L’existence même d’une HTP impacte directement le pronostic du patient BPCO : majoration des symptômes et du taux d’hospitalisation, baisse de la survie. La survie à quatre ans d’un patient ayant une BPCO est de près de 75 %, tous stades confondus, mais lorsque la BPCO est associée à une HTP, elle chute à 50 %.4
Concernant les médicaments approuvés pour le traitement de l’HTAP dans la BPCO :6,7 les études concernent de petits effectifs, sont de durée courte, avec des données hémodynamiques incomplètes et surtout des résultats discordants, avec de possibles effets délétères. Par conséquent, ces traitements ne sont pas recommandés pour traiter l’HTP du groupe 3. Néanmoins, en cas d’HTP sévère, l’inclusion des patients dans de larges études randomisées s’avère nécessaire.
La prise en charge de ces patients atteints de BPCO avec HTP reste individualisée et du ressort des centres experts. Devant l’absence de recommandation pour ou contre l’utilisation des inhibiteurs de la phosphodiestérase de type 5, cette classe médicamenteuse est parfois prescrite.
Concernant les médicaments approuvés pour le traitement de l’HTAP dans la BPCO :6,7 les études concernent de petits effectifs, sont de durée courte, avec des données hémodynamiques incomplètes et surtout des résultats discordants, avec de possibles effets délétères. Par conséquent, ces traitements ne sont pas recommandés pour traiter l’HTP du groupe 3. Néanmoins, en cas d’HTP sévère, l’inclusion des patients dans de larges études randomisées s’avère nécessaire.
La prise en charge de ces patients atteints de BPCO avec HTP reste individualisée et du ressort des centres experts. Devant l’absence de recommandation pour ou contre l’utilisation des inhibiteurs de la phosphodiestérase de type 5, cette classe médicamenteuse est parfois prescrite.
Lire aussi | Hypertension pulmonaire : les 10 messages-clés
Particularités de l’HTP des pneumopathies interstitielles diffuses (groupe 3.2)
La prévalence de l’HTP dans les cohortes de patients suivis pour une fibrose pulmonaire idiopathique est de 8 à 15 % chez les patients nouvellement diagnostiqués, de 30 à 50 % chez les patients ayant une atteinte parenchymateuse modérée et de plus de 80 % dans les formes évoluées de la maladie.8,9
Le diagnostic d’HTP lors du suivi évolutif d’une PID marque un tournant évolutif de la maladie et une évolution péjorative à court terme. L’HTP dans ce contexte a un moins bon pronostic que lorsqu’elle est associée à une BPCO.10 Le registre européen COMPERA a mis en évidence une médiane de survie de 1,8 [1,6-2] ans après le diagnostic d’HTP.11
Le traitement est avant tout celui de la maladie causale (traitement antifibrosant ou traitement immunosupresseur en fonction de la cause de la PID) ; compte tenu du pronostic, le recours à une transplantation bipulmonaire doit être envisagé pour les patients éligibles. Selon les recommandations européennes en cours, il est conseillé d’adresser les patients à un centre de compétence ou de référence de l’HTP, en cas de forme sévère uniquement, afin qu’une approche thérapeutique individualisée soit proposée.
Comme dans la BPCO, les inhibiteurs de la phosphodiestérase de type 5 sont parfois prescrits. Un traitement par tréprostinil inhalé peut être envisagé chez les patients atteints d’HTP associée à une PID.
L’ambrisentan et le riociguat sont contre-indiqués.1
Le diagnostic d’HTP lors du suivi évolutif d’une PID marque un tournant évolutif de la maladie et une évolution péjorative à court terme. L’HTP dans ce contexte a un moins bon pronostic que lorsqu’elle est associée à une BPCO.10 Le registre européen COMPERA a mis en évidence une médiane de survie de 1,8 [1,6-2] ans après le diagnostic d’HTP.11
Le traitement est avant tout celui de la maladie causale (traitement antifibrosant ou traitement immunosupresseur en fonction de la cause de la PID) ; compte tenu du pronostic, le recours à une transplantation bipulmonaire doit être envisagé pour les patients éligibles. Selon les recommandations européennes en cours, il est conseillé d’adresser les patients à un centre de compétence ou de référence de l’HTP, en cas de forme sévère uniquement, afin qu’une approche thérapeutique individualisée soit proposée.
Comme dans la BPCO, les inhibiteurs de la phosphodiestérase de type 5 sont parfois prescrits. Un traitement par tréprostinil inhalé peut être envisagé chez les patients atteints d’HTP associée à une PID.
L’ambrisentan et le riociguat sont contre-indiqués.1
Particularités de l’HTP du syndrome emphysème-fibrose (groupe 3.3)
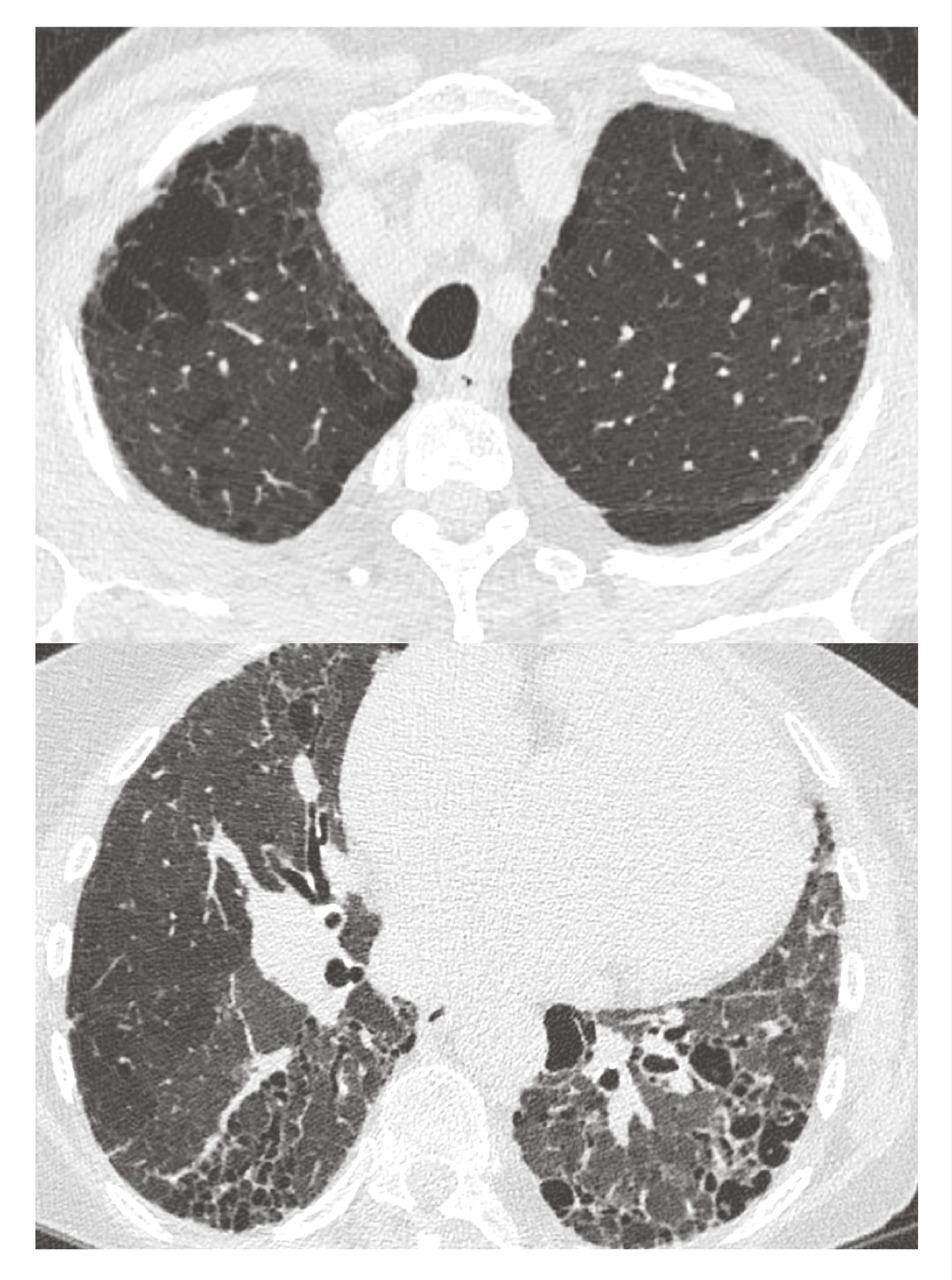
Le syndrome emphysème-fibrose est défini par la coexistence de lésions d’emphysème aux sommets et d’une fibrose pulmonaire aux bases. La prévalence de l’HTP est particulièrement élevée dans ce contexte (de 15 à 55 % des patients). Cette association comorbide est responsable d’une diminution de la DLCO et d’une hypoxémie sévère de très mauvais pronostic quel que soit le traitement (figure ).
Lire aussi | Dépistage et diagnostic de l’hypertension pulmonaire
Particularités de l’HTP des hypoventilations alvéolaires chroniques (groupe 3.4)
La cause actuelle la plus fréquente en France d’hypoventilation alvéolaire est le syndrome obésité-hypoventilation. Il se complique très fréquemment d’une HTP parfois sévère. Il est important d’y penser chez une personne obèse ayant une dyspnée d’effort importante et des œdèmes des membres inférieurs.
Des explorations fonctionnelles respiratoires comprenant des gaz du sang artériel (idéalement réalisés au réveil) et un enregistrement du sommeil permettent de parvenir au diagnostic.
Le traitement est fondé sur la pression positive continue (PPC) ou la ventilation non invasive nocturne, qui permettent de corriger l’hypoventilation alvéolaire et d’améliorer de manière significative l’HTP.13
Des explorations fonctionnelles respiratoires comprenant des gaz du sang artériel (idéalement réalisés au réveil) et un enregistrement du sommeil permettent de parvenir au diagnostic.
Le traitement est fondé sur la pression positive continue (PPC) ou la ventilation non invasive nocturne, qui permettent de corriger l’hypoventilation alvéolaire et d’améliorer de manière significative l’HTP.13
Lire aussi | Hypertension pulmonaire thromboembolique chronique
Évolution de la classification
La nouvelle classification présentée au 7e Congrès mondial, en juin 2024 (World Symposium on Pulmonary Hypertension),14 propose de ne plus seulement se fonder sur la fonction respiratoire mais aussi sur des diagnostics cliniques, soulignant l’importance de l’imagerie (tableau 2 ). Ainsi la BPCO et/ou l’emphysème, les pneumopathies interstitielles diffuses, les syndromes emphysème-fibrose ou encore les hypoventilations alvéolaires sont bien identifiés pour une prise en charge plus personnalisée de pathologies à pronostics différents et prenant en compte les mécanismes physiopathologiques et la présentation clinique.
L’importance de réussir à mieux phénotyper l’ensemble des malades avec HTP associée aux maladies respiratoires chroniques est également soulignée du fait de la difficulté à les prendre en charge.
L’importance de réussir à mieux phénotyper l’ensemble des malades avec HTP associée aux maladies respiratoires chroniques est également soulignée du fait de la difficulté à les prendre en charge.
Prendre d’abord en charge la pathologie respiratoire causale
L’HTP associée aux maladies respiratoires chroniques (groupe 3) est fréquente et de mauvais pronostic. La prise en charge passe tout d’abord par celle de la maladie respiratoire causale. Les dossiers des patients ayant une HTP sévère (RVP > 5 UW) doivent être discutés de manière individualisée avec l’équipe du centre de compétence ou de référence de l’HTP, puis, si nécessaire, le patient est adressé pour débattre de la mise en place de thérapeutiques spécifiques.
Références
1. Humbert M, Kovacs G, Hoeper MM, et al. ESC/ERS Scientific Document Group. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 2023;61(1):2200879.
2. Hurdman J, Condliffe R, Elliot CA, et al. ASPIRE registry: Assessing the Spectrum of Pulmonary hypertension Identified at a REferral centre. Eur Respir J 2012;39(4):945-55.
3. Chaouat A, Bugnet AS, Kadaoui N, et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2005;172(2):189-94.
4. Kovacs G, Avian A, Bachmaier G, et al. Severe pulmonary hypertension in COPD: Impact on survival and diagnostic approach. Chest 2022;162(1):202-12.
5. Rahaghi FF, Kolaitis NA, Adegunsoye A, et al. Screening strategies for pulmonary hypertension in patients with interstitial lung disease: A multidisciplinary delphi study. Chest 2022;162(1):145‑55.
6. Hao Y, Zhu Y, Mao Y, et al. Efficacy and safety of sildenafil treatment in pulmonary hypertension caused by chronic obstructive pulmonary disease: A meta-analysis. J Life Sci 2020;257:118001.
7. Vitulo P, Stanziola A, Confalonieri M, et al. Sildenafil in severe pulmonary hypertension associated with chronic obstructive pulmonary disease: A randomized controlled multicenter clinical trial. J Heart Lung Transplant 2017;36(2):166-74.
8. Nadrous HF, Pellikka PA, Krowka MJ, et al. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Chest 2005;128(4):2393‑9.
9. Nathan SD, Shlobin OA, Ahmad S, et al. Serial development of pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respir Int Rev Thorac Dis 2008;76(3):288‑94.
10. Chebib N, Mornex JF, Traclet J, et al. Pulmonary hypertension in chronic lung diseases: Comparison to other pulmonary hypertension groups. Pulm Circ 2018;8(2):2045894018775056.
11. Hoeper MM, Behr J, Held M, et al. Pulmonary hypertension in patients with chronic fibrosing idiopathic interstitial pneumonias. PloS One 2015;10(12):e0141911.
12. Cottin V, Selman M, Inoue Y, et al. Syndrome of combined pulmonary fibrosis and emphysema: An official ATS/ERS/JRS/ALAT research statement. Am J Respir Crit Care Med 2022;206(4):e7‑41.
13. Adir Y, Humbert M, Chaouat A. Sleep-related breathing disorders and pulmonary hypertension. Eur Respir J 2021;57(1):2002258.
14. Shlobin OA, Adir Y, Barbera JA, et al. Pulmonary hypertension associated with lung diseases. Eur Respir J 2024;64(4):2401200.
2. Hurdman J, Condliffe R, Elliot CA, et al. ASPIRE registry: Assessing the Spectrum of Pulmonary hypertension Identified at a REferral centre. Eur Respir J 2012;39(4):945-55.
3. Chaouat A, Bugnet AS, Kadaoui N, et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2005;172(2):189-94.
4. Kovacs G, Avian A, Bachmaier G, et al. Severe pulmonary hypertension in COPD: Impact on survival and diagnostic approach. Chest 2022;162(1):202-12.
5. Rahaghi FF, Kolaitis NA, Adegunsoye A, et al. Screening strategies for pulmonary hypertension in patients with interstitial lung disease: A multidisciplinary delphi study. Chest 2022;162(1):145‑55.
6. Hao Y, Zhu Y, Mao Y, et al. Efficacy and safety of sildenafil treatment in pulmonary hypertension caused by chronic obstructive pulmonary disease: A meta-analysis. J Life Sci 2020;257:118001.
7. Vitulo P, Stanziola A, Confalonieri M, et al. Sildenafil in severe pulmonary hypertension associated with chronic obstructive pulmonary disease: A randomized controlled multicenter clinical trial. J Heart Lung Transplant 2017;36(2):166-74.
8. Nadrous HF, Pellikka PA, Krowka MJ, et al. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Chest 2005;128(4):2393‑9.
9. Nathan SD, Shlobin OA, Ahmad S, et al. Serial development of pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respir Int Rev Thorac Dis 2008;76(3):288‑94.
10. Chebib N, Mornex JF, Traclet J, et al. Pulmonary hypertension in chronic lung diseases: Comparison to other pulmonary hypertension groups. Pulm Circ 2018;8(2):2045894018775056.
11. Hoeper MM, Behr J, Held M, et al. Pulmonary hypertension in patients with chronic fibrosing idiopathic interstitial pneumonias. PloS One 2015;10(12):e0141911.
12. Cottin V, Selman M, Inoue Y, et al. Syndrome of combined pulmonary fibrosis and emphysema: An official ATS/ERS/JRS/ALAT research statement. Am J Respir Crit Care Med 2022;206(4):e7‑41.
13. Adir Y, Humbert M, Chaouat A. Sleep-related breathing disorders and pulmonary hypertension. Eur Respir J 2021;57(1):2002258.
14. Shlobin OA, Adir Y, Barbera JA, et al. Pulmonary hypertension associated with lung diseases. Eur Respir J 2024;64(4):2401200.
Dans cet article
- Physiopathologie mal élucidée
- Poser le diagnostic
- Approche thérapeutique globale
- Particularités de l’HTP sur la BPCO (groupe 3.1)
- Particularités de l’HTP des pneumopathies interstitielles diffuses (groupe 3.2)
- Particularités de l’HTP du syndrome emphysème-fibrose (groupe 3.3)
- Particularités de l’HTP des hypoventilations alvéolaires chroniques (groupe 3.4)
- Évolution de la classification
- Prendre d’abord en charge la pathologie respiratoire causale

Résumé
L’hypertension pulmonaire est fréquemment observée au cours des maladies respiratoires chroniques. Elle est responsable d’une détérioration de la qualité de vie et d’une diminution de la survie des patients à court et à moyen termes. Comme dans toutes les autres formes d’hypertension pulmonaire, le diagnostic formel repose sur les données du cathétérisme cardiaque droit. Elle est dite sévère si les résistances vasculaires pulmonaires sont supérieures à 5 unités Wood ; dans ce cas uniquement, une prise en charge spécifique doit être discutée au sein d’un centre de compétence ou de référence des maladies vasculaires pulmonaires.