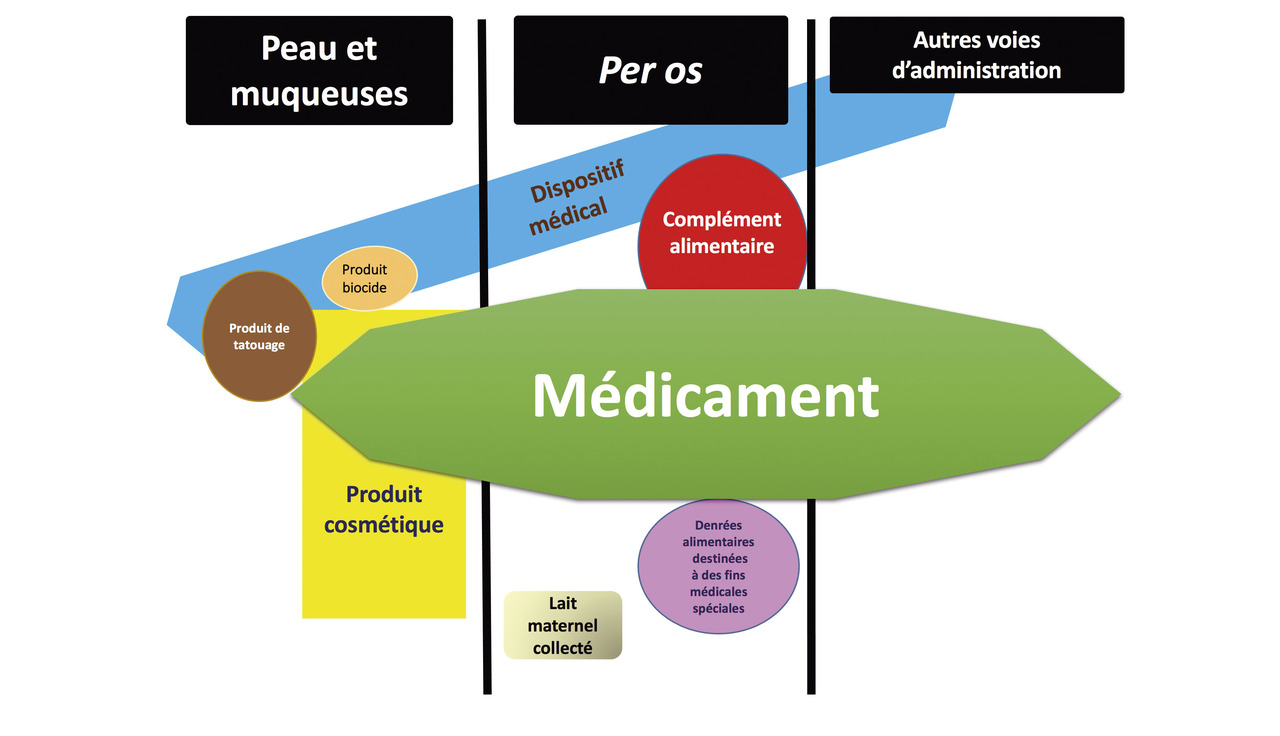
Lister les différents types de produits qui sont l’objet d’une vigilance réglementée.
Discuter une situation où la vigilance pourrait être déficitaire pour chacun de ces produits.
Préciser les grandes missions de Santé publique France et de l’ANSM.
Le médicament
- la fabrication, la distribution et la dispensation des médicaments sont réservées aux pharmaciens (monopole pharmaceutique) ;
- l’accès du patient au médicament est très souvent subordonné à la prescription d’un médecin – médicaments soumis à la réglementation des substances vénéneuses (liste I, liste II, stupéfiants) –, voire à certaines catégories de médecins (hospitaliers, spécialistes, etc.).
Typologie des risques liés au médicament (tableau 1)
1) Produit : bien matériel, objet tangible (gélule, ampoule injectable, collyre…), destiné à être consommé. La qualité du produit est la conformité à la formule annoncée, garantie par les procédés de fabrication industrielle.
2) Information : bien immatériel, ensemble des données relatives à la qualité du produit, à sa sécurité, à son efficacité contenues dans le dossier d’AMM.
3) Usage : par les professionnels de santé et le patient.
Ces trois éléments, très encadrés par des règles du code de la santé publique, sont étroitement intriqués.
Risques liés à la qualité du produit
Le défaut de qualité du produit peut être :- soit involontaire, résultant d’une faille dans le circuit légal du médicament, une malfaçon qui a échappé à la vigilance des professionnels ;
- soit volontaire, avec les médicaments falsifiés, la tromperie pouvant concerner l’objet matériel et/ou l’information associée.
Risques liés à l’information sur le produit
À cause du caractère expérimental des essais cliniques – par construction, toujours trop courts, trop étroits et réalisés sur des populations peu représentatives –, de la complexité de l’évaluation du médicament et de la quantification des risques, les connaissances acquises sur le rapport bénéfice/risque du médicament lors de sa commercialisation sont toujours très limitées. De nouveaux risques peuvent être identifiés sur une population plus large ; on peut les différencier selon un gradient de prévisibilité et d’évitabilité en trois groupes, que nous illustrerons par des exemples de médicaments qui ont donné lieu à des procès en responsabilité :- l’ignorance des risques, en l’absence de données toxicologiques ; par exemple, le défaut d’information sur les effets toxiques du médicament pour l’embryon ou le fœtus : tératogénicité de la thalidomide, fœtoxicité du valproate de sodium (Depakine), méconnaissance du caractère transmissible d’effets nocifs sur plusieurs générations du diéthylstilbestrol (Distilbène) ;
- le défaut d’expertise de l’information : faute d’analyse approfondie des données de la recherche clinique, des effets indésirables qui auraient pu être identifiés ne l’ont pas été. Les négligences peuvent provenir du demandeur de l’AMM, des instances d’évaluation, ou des deux à la fois, en raison de liens d’intérêts entre laboratoire et autorités sanitaires ; par exemple, Vioxx (rofécoxib) et Mediator (benfluorex) ;
- des informations falsifiées, issues d’actes délibérément frauduleux de la part d’acteurs de la chaîne du médicament.
Risques liés à l’usage
Par ailleurs, un médicament sûr mais mal prescrit, mal dispensé ou mal utilisé peut aussi entraîner de graves dommages. Le caractère inapproprié de l’usage du médicament peut résulter de sa prescription, de sa dispensation ou de son administration, faisant intervenir, selon les cas, le prescripteur, le pharmacien, l’infirmier et, finalement, le patient lui-même. À titre d’exemple, on peut citer les malaises et les fausses routes de nourrissons et le décès d’un nouveau-né après la prise orale de vitamine D (Uvestérol). Ces tristes événements ont montré comment les modalités d’administration par l’utilisateur final peuvent amener à suspendre la commercialisation d’un médicament irréprochable quant à sa qualité et à l’information associée9.Plus généralement, deux types de risques liés à l’usage doivent être distingués :
- l’utilisation inappropriée non intentionnelle, qualifiée d’erreur médicamenteuse (v. item 325). D’après les dernières enquêtes, un événement indésirable grave évitable dû à un médicament se produirait dans les établissements de santé pour 2 000 journées d’hospitalisation, soit plus de 50 000 événements graves évitables par an en France10 ;
- l’utilisation inappropriée intentionnelle, qualifiée de mésusage. La fréquence des prescriptions hors AMM est estimée à 20 % et apparaît plus élevée en pédiatrie, psychiatrie et oncologie. Dans l’affaire du Mediator, cette proportion a atteint 70 %. Ces mésusages sont associés à des risques imprévisibles, faute de données cliniques. Rappelons cependant qu’en l’absence d’alternative médicamenteuse autorisée, les prescriptions hors AMM sont licites ; elles sont laissées à la discrétion et responsabilité du seul prescripteur.
Politique de sécurité sanitaire en matière de médicament
Recherche de la sécurité des produits
La démarche de gestion des risques de l’ANSM est organisée autour de plusieurs axes.Recherche de la qualité du produit
- à travers les inspections : audits des pharmaciens inspecteurs sur le site des établissements pharmaceutiques de fabrication et de distribution pour vérifier la conformité des activités aux référentiels (bonnes pratiques de fabrication, préconisations du dossier d’AMM, bonnes pratiques de distribution…) ;
- à travers les contrôles en laboratoire : analyse de la composition d’échantillons de médicaments.
Recherche de la sécurité du produit autorisé, à travers les missions de surveillance
Le système de pharmacovigilance vise à s’assurer du maintien de l’acceptabilité du rapport bénéfice/risque de tout médicament après sa mise sur le marché.
De plus, pour ceux jugés sensibles, des mesures complémentaires sont mises en place : une surveillance renforcée décidée au niveau de l’Union européenne (UE) pour une période de 5 ans (triangle noir inversé sur le résumé des caractéristiques du produit et sur la notice), un plan de gestion des risques (PGR), décidé au niveau européen ou national, pour mieux caractériser et quantifier les risques, obtenir des informations manquantes ou surveiller le bon usage dans les conditions réelles d’utilisation ; avec le contrôle de la publicité, qu’elle soit destinée aux professionnels de santé ou au grand public.
Mesures de police sanitaire
Recherche de la sécurité du patient
L’ANSM s’est mobilisée face aux risques d’usage inapproprié avec toute une série d’initiatives :- minimisation des risques de mésusage, avec la création d’un observatoire des consommations anormales, sur le plan quantitatif ou qualitatif ;
- identification des erreurs médicamenteuses afin de les prévenir ;
. échanges de l’ANSM avec les médecins pour partager les expériences et identifier les conditions liées aux plus grands risques d’erreurs ;
. mesures de réduction des risques : demande de modification de l’AMM, de la notice ou des conditionnements, communication auprès des professionnels ou du public, amélioration et harmonisation de l’étiquetage, recommandations et campagnes d’information concernant les dispositifs d’administration, etc. ;
- information des patients sur les risques particuliers associés à certains médicaments sensibles : pictogrammes grossesse pour les médicaments présentant des risques pour les femmes enceintes, messages d’alerte sur le risque de toxicité hépatique apposés sur les conditionnements de spécialités de paracétamol, etc.
Autres produits réglementés
Comme pour le médicament, en plus des éventuels effets indésirables ou risques d’incidents annoncés par l’étiquetage du produit, des risques inattendus peuvent survenir, liés à la non-qualité du produit, à son information ou à son usage.
Dispositifs médicaux, dispositifs médicaux de diagnostic in vitro et produits cosmétiques : sous tutelle de l’ANSM
Elle peut décider des retraits de produits et toute autre mesure de police sanitaire.
Denrées alimentaires destinées à des fins médicales spéciales, compléments alimentaires (CA) et produits biocides
La surveillance après commercialisation des deux catégories de produits alimentaires particuliers (DADFMS, CA) est confiée à l’ANSES, responsable de la nutrivigilance.
Les retraits de produits du marché, les interdictions de commercialisation et toute autre mesure de police sanitaire de ces trois catégories sont pris, comme pour les produits de consommation ordinaire, par la Direction générale de la concurrence, de la consommation et de la répression des fraudes (DGCCRF).
La surveillance des produits biocides est celle de tout produit de consommation ordinaire : le dispositif de toxicovigilance, avec le réseau des centres antipoison.
La sécurité sanitaire passe par la mobilisation de tous les professionnels de santé, qui doivent signaler les effets indésirables ou événements graves susceptibles d’être dus à ces produits réglementés16. Pour faciliter les déclarations des professionnels et des patients, il existe un portail national de signalement de tous les événements sanitaires indésirables. Pour plusieurs catégories de produits, les déclarations peuvent se faire en outre directement via le site de l’agence correspondante (ANSM, ANSES).
Enfin, la promotion de la sécurité sanitaire est également poursuivie à travers certaines missions de la HAS et des ARS. Au niveau régional, la coordination de l’ensemble des dispositifs de vigilance est assurée par un réseau régional de vigilances et d'appui (RREVA).
POINTS FORTS À RETENIR
Bien différencier les dispositifs de vigilance en fonction du statut des produits auxquels ils s’appliquent.
Bien distinguer les risques connus de ceux qui s’avèrent inattendus et relèvent soit de la non-qualité du produit (non-conformité à la formule annoncée), soit de l’information associée, soit de l’usage qui en fait.
Savoir définir les notions de malfaçon, de falsification, d’erreur médicamenteuse et de mésusage.
Connaître les missions de l’ANSM en matière de sécurité sanitaire des produits (expertise scientifique, inspection et contrôles, vigilance, police sanitaire) et des patients (observatoire des consommations, signalement des erreurs médicamenteuses, pictogrammes d’information des patients).
Savoir retrouver les données disponibles sur le site de l’ANSM : « Informations de sécurité », « Décisions » et savoir identifier les mesures de police sanitaire.
Pour en savoir +
La sécurité sanitaire. Tabuteau D. 2e éd. Berger-Levrault, 2002.
La sécurité sanitaire des produits destinés à l’homme. La veille sanitaire (voir item 325)
Cet item transversal peut faire l’objet de questions à choix simple ou multiple, dans le cadre d’un dossier ou sous forme d’une question isolée de connaissance générale. Par exemple, la question suivante peut être envisageable :
Parmi les propositions suivantes, laquelle (lesquelles) est(sont) exacte(s) ?
A. Un défaut de qualité involontaire peut être qualifié de falsification
B. Une utilisation inappropriée non intentionnelle d’un médicament peut être qualifiée de mésusage
C. En matière de médicaments, l’ANSM peut inspecter un établissement de fabrication, contrôler la publicité et décider d’une suspension d’AMM
D. Le dispositif de vigilance applicable à une bandelette urinaire est la réactovigilance
E. L’ANSM est compétente pour retirer du marché un complément alimentaire pour des raisons de sécurité.
 Encadrés
Encadrés