La bile, synthétisée par le foie, joue un rôle essentiel dans la digestion des grais-ses, l’absorption intestinale des lipides et l’élimination de substances toxiques. La sécrétion par les hépatocytes de ses principaux composés organiques, en particulier des acides biliaires, du cholestérol et des phospholipides, dépend de transporteurs spécifiques situés au pôle canaliculaire de ces cellules. L’activité des transporteurs biliaires est étroitement régulée afin de maintenir l’homéostasie biliaire.
La fluidité, ou à l’inverse la viscosité, de la bile est liée à sa composition et à la proportion de ses différents composants organiques. En cas d’excès ou de défaut d’un composant biliaire, la capacité de solubilisation du cholestérol (composant hydrophobe) peut être altérée et entraîner sa précipitation sous forme de concrétions, ou calculs biliaires.
Physiopathologie du syndrome LPAC
Dans la lithiase biliaire commune, la formation de calculs est liée à une sursaturation de la bile en cholestérol, situation fréquemment observée en cas de surpoids ou chez le sujet âgé. Dans ce cas, le cholestérol précipite principalement dans la vésicule biliaire, où la bile a tendance à stagner entre les périodes perprandiales.
La lithiase biliaire par déficit en phospholipides, ou syndrome LPAC (low phospholipid-associated cholelithiasis), est une forme génétique rare de lithiase biliaire intrahépatique due à une faible concentration biliaire en phospholipides, à l’origine d’une perte de solubilisation du cholestérol. Ce syndrome a été décrit pour la première fois en 2001 par l’équipe du Pr Poupon, de l’hôpital Saint-Antoine à Paris.1 Il a été originellement associé à l’existence de variants pathogènes du gène ABCB4 (ATP binding cassette subfamily B member 4) codant pour le transporteur des phospholipides MDR3 (multidrug resistance protein 3), qui est situé au pôle apical (canaliculaire) des hépatocytes. La diminution de la sécrétion biliaire en phospholipides induit un défaut de formation des micelles mixtes (associant cholestérol, acides biliaires et phospholipides) dans la bile, qui sont les principaux vecteurs de solubilisation du cholestérol biliaire. Le cholestérol non solubilisé précipite ainsi dans les canalicules et les canaux biliaires intrahépatiques sous forme de microcristaux, dont l’agrégation aboutit à la formation de calculs.
Caractérisation génétique
Le transporteur MDR3 est codé par le gène ABCB4 localisé au niveau du locus 21 du chromosome 7 (7q21). Un variant pathogène du gène ABCB4 n’est retrouvé que chez 30 à 50 % des patients présentant un phénotype clinique de syndrome LPAC (voir plus loin les critères diagnostiques du syndrome), ce qui souligne l’hétérogénéité physiopathologique de cette entité et l’implication plus que probable d’autres facteurs pathogènes, qu’ils soient génétiques (constitutionnels) ou environnementaux.2 La pénétrance du syndrome LPAC est incomplète (inférieure à 50 %) et la plupart des cas rapportés sont sporadiques. Quand un variant pathogène du gène ABCB4 est identifié, il s’agit dans deux tiers des cas d’un variant faux-sens hétérozygote.3,4 Des variants faux-sens bialléliques, des variants non-sens, des variants d’épissage ou des délétions/insertions avec décalage du cadre de lecture sont plus rarement observés. Une classification des variants pathogènes du gène ABCB4 permet de distinguer les variants majeurs (non-sens, décalage du cadre de lecture) dits de classe I, associés à une perte de la protéine, des variants faux-sens responsables d’un défaut de maturation (classe II), d’un défaut d’activité (classe III), d’un défaut de stabilité (classe IV) ou d’un défaut d’un partenaire d’interaction (classe V) de la protéine MDR3.5 Cette classification pourrait avoir un intérêt pour le développement de molécules thérapeutiques. Un déficit complet du gène ABCB4 est associé à une maladie hépatique sévère (cirrhose biliaire) de l’enfant, nommée cholestase intrahépatique familiale progressive de type 3.
Données épidémiologiques
Le syndrome LPAC est diagnostiqué dans la majorité des cas chez des adultes jeunes, avec un âge médian de 36 ans au moment du diagnostic. L’âge médian au moment des premiers symptômes, quant à lui, n’est que de 27 ans, ce qui souligne un retard diagnostique souvent important. La maladie est exceptionnelle chez le jeune enfant, mais peut se révéler chez l’adolescent. Des cas de révélation tardive sont parfois observés (15 % des cas sont découverts après 40 ans). Il existe une nette prédominance féminine (3 femmes pour 1 homme), confirmant le rôle prédisposant du sexe féminin vis-à-vis du risque de lithiase biliaire en général et l’effet inhibiteur des hormones sexuelles féminines sur l’expression des transporteurs biliaires en particulier. Dans ce contexte, il faut noter la forte prévalence de la cholestase gravidique chez les femmes atteintes du syndrome LPAC (de l’ordre de 40 % contre 2 % pour la lithiase biliaire classique).
Le syndrome LPAC est estimé responsable d’environ 1 % de l’ensemble des cas de lithiase biliaire symptomatique de l’adulte.4 Sa prévalence en population générale se situerait entre 10 et 40 pour 100 000 habitants. Cependant, chez les patients de moins de 30 ans présentant une lithiase biliaire symptomatique, la fréquence du syndrome LPAC pourrait dépasser 25 %.6 - 8
Présentations clinique et radiologique
Signes cliniques de lithiase biliaire à un âge jeune
La présentation clinique du syndrome LPAC rejoint celle de la lithiase biliaire dite « classique » et se caractérise par la survenue de crises douloureuses de colique hépatique, associées parfois à des complications biliaires à type de lithiase de la voie biliaire principale, d’angiocholite ou de pancréatite aiguë lithiasique. De manière caractéristique, les douleurs biliaires surviennent chez des patients de moins de 40 ans sans facteurs de risque habituels de lithiase biliaire (absence de surpoids et de dyslipidémie). La récidive des symptômes biliaires après cholécystectomie est très évocatrice du syndrome LPAC et constitue l’un des critères diagnostiques forts. Un antécédent de lithiase biliaire chez les apparentés du premier degré est retrouvé chez 48 % des patients (contre 34 % dans la lithiase biliaire classique). En dehors des épisodes de migration lithiasique qui peuvent accompagner les crises avec augmentation aiguë transitoire des transaminases, la biochimie hépatique est le plus souvent normale. Une augmentation chronique des gamma GT peut être observée (dans un tiers des cas environ), plus particulièrement chez les patients porteurs d’un variant pathogène du gène ABCB4.4
Échographie hépatique : examen clé du diagnostic
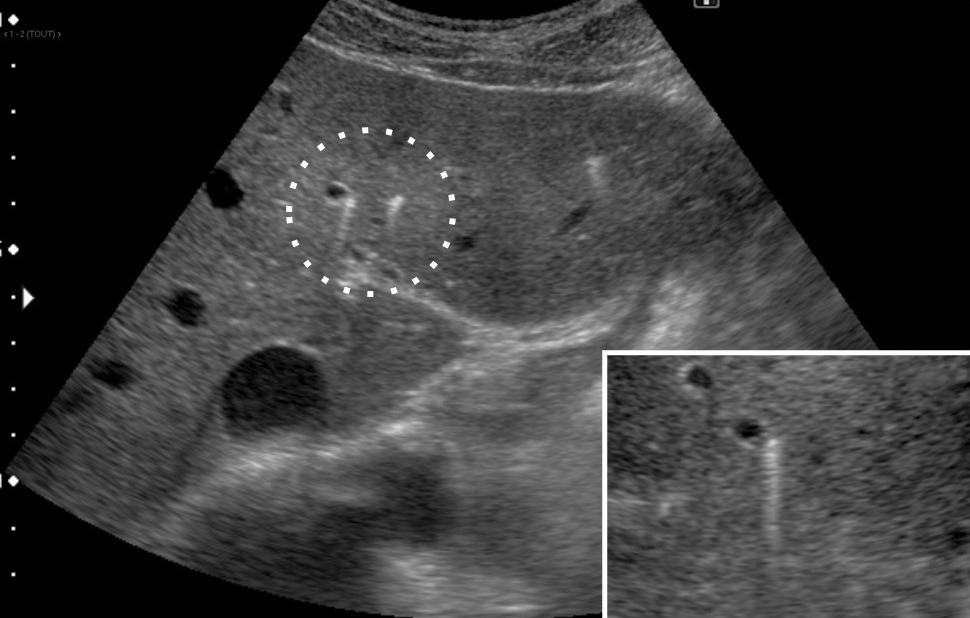
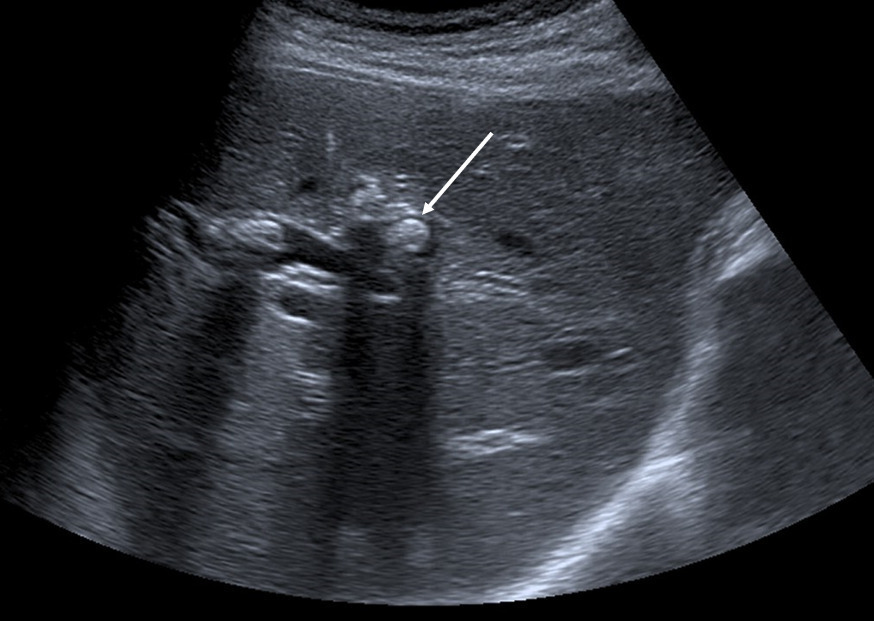
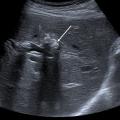
L’échographie hépatique est l’examen clé pour le diagnostic de syndrome LPAC. Celle-ci met typiquement en évidence les signes d’une microlithiase intrahépatique sous la forme d’images en « queue de comète » (échos de répétition liés à la vibration des microcristaux sous le faisceau d’ultrasons), de micro-spots ou de « boue biliaire » (sludge) le long des canaux intrahépatiques (fig. 1A). Ces signes sont parfois difficilement identifiables, notamment en situation non sensibilisée et pour un opérateur non entraîné. Le principal diagnostic différentiel est représenté par les microhamartomes (ou complexes de von Meyenburg) qui peuvent parfois se présenter sous la forme d’artéfacts en queue de comète.9 Une échographie normale standard n’élimine pas le diagnostic et il peut être nécessaire de répéter l’examen par un radiologue expérimenté et formé à la sémiologie du syndrome LPAC. Moins fréquemment (5 à 10 % des cas), le syndrome LPAC peut se présenter sous la forme de vrais calculs (macrolithiases) intra- ou extrahépatiques (fig. 1B).4 Une lithiase vésiculaire n’est observée que chez un tiers des patients atteints de syndrome LPAC.
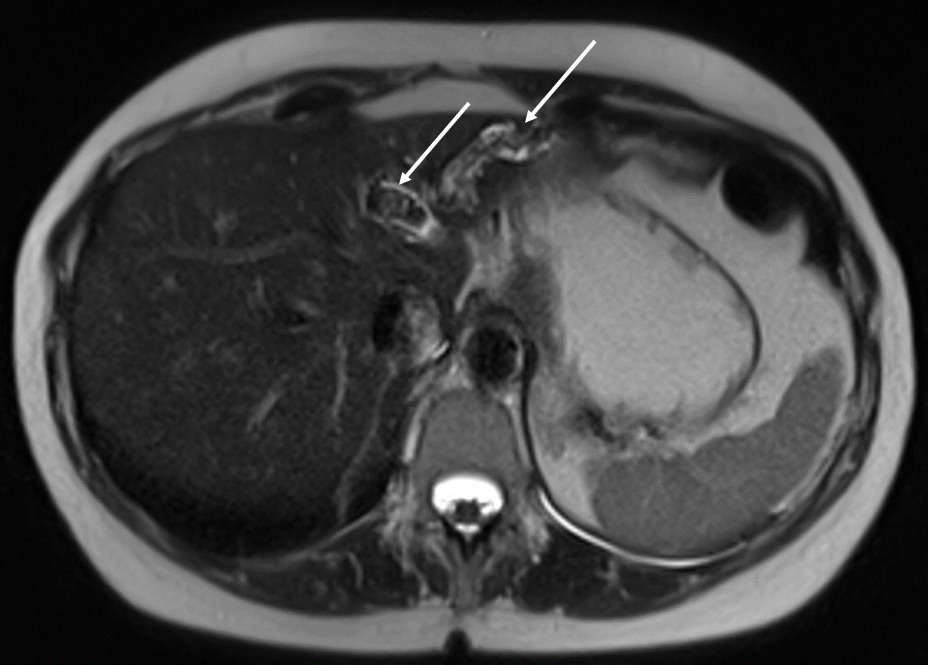
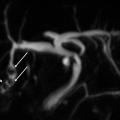
La cholangiographie par IRM est typiquement normale dans les formes habituelles, microlithiasiques, du syndrome LPAC. Elle n’est donc pas nécessaire au diagnostic dans la très grande majorité des situations. Elle doit être réalisée en cas de calculs intrahépatiques ou de dilatation des canaux biliaires en échographie afin d’évaluer le retentissement de ces calculs sur les voies biliaires et le parenchyme hépatique. Les calculs biliaires ne sont pas toujours visibles à l’imagerie par résonance magnétique (IRM), notamment quand leur taille est inférieure à 5 mm. Lorsqu’ils sont visibles, ils sont le plus souvent visualisés en hyposignal T1 et T2 sous forme de lacunes endobiliaires (fig. 2 A et 2B). Dans une série récente portant sur 125 patients suivis dans un centre expert et ayant tous eu une bili-IRM, 49 % présentaient des anomalies des canaux biliaires, parmi lesquelles 93 % de calculs intrahépatiques et 38 % de dilatation des canaux biliaires.10 Ces anomalies morphologiques étaient plus fréquemment observées chez les porteurs d’un variant pathogène du gène ABCB4.
Diagnostic positif
Le diagnostic de syndrome LPAC repose actuellement sur la présence d’au moins deux critères diagnostiques « majeurs » sur trois (tableau)parmi les suivants :1
- début de la symptomatologie biliaire avant 40 ans ;
- récidive des symptômes biliaires après cholécystectomie ;
- signes de (micro)lithiase intra-hépatique en échographie.
Des critères diagnostiques « mineurs » peuvent également aider au diagnostic de syndrome LPAC. La présence d’antécédents personnels ou familiaux de cholestase gravidique ou d’antécédents familiaux de lithiase biliaire du sujet jeune (moins de 40 ans) peuvent renforcer cette hypothèse.4 Ainsi, si un seul critère majeur de LPAC est retrouvé, la présence d’au moins deux critères mineurs associés peut permettre de poser le diagnostic. L’absence de surpoids et l’efficacité clinique (a posteriori) du traitement par l’acide ursodésoxycholique (AUDC) sont également des facteurs qui peuvent être pris en compte.
La recherche de variants pathogènes du gène ABCB4, réalisée par séquençage à haut débit (NGS, next generation sequencing), n’est pas nécessaire au diagnostic clinique dans la mesure où elle n’est positive que chez 30 à 50 % des patients ayant un phénotype clinique caractéristique de syndrome LPAC et que son résultat ne modifie pas la prise en charge. Elle n’est d’ailleurs pas remboursée par la Sécurité sociale et doit être réalisée si possible dans un centre expert. Sa valeur n’a d’intérêt que positive, en confirmant le diagnostic sur le plan moléculaire et en facilitant le dépistage intrafamilial. Si cette recherche génétique est faite, elle ne doit en aucun cas retarder la mise en route du traitement, car le délai d’obtention des résultats est souvent de plusieurs mois.
La recherche de variants du gène ABCB4 présente un intérêt en recherche clinique, en particulier pour améliorer la caractérisation clinique et génétique du syndrome. À cet égard, certaines particularités phénotypiques du syndrome semblent dépendre du génotype d’ABCB4 (encadré).
Complications lithiasiques du syndrome LPAC
Les complications du syndrome LPAC sont celles de la lithiase biliaire en général, à savoir principalement l’obstruction aiguë de la voie biliaire principale par migration lithiasique, l’angiocholite et la pancréatite aiguë lithiasique.4 La fréquence des angiocholites aiguës est plus élevée dans le syndrome LPAC (24 %) que dans la lithiase biliaire classique (7 %). En revanche, le risque de cholécystite aiguë est moins important que dans la lithiase classique.4
Dans les rares formes macrolithiasiques, des signes radiologiques de cholangite sclérosante sont parfois décrits, et l’évolution vers une cirrhose biliaire secondaire, bien que rare, est alors possible. Dans ces formes sévères, il existe un risque faible mais significatif de cholangiocarcinome, notamment chez les patients porteurs d’un variant pathogène du gène ABCB4. Dans une série multicentrique française portant sur 308 patients atteints de syndrome LPAC, cinq cas de cholangiocarcinome intrahépatique ont été rapportés.4 Ces cholangiocarcinomes sont survenus chez des patients ayant des calculs intrahépatiques symptomatiques compliqués de dilatation des canaux biliaires et porteurs d’un variant ABCB4. Même si ces cas sont rares, ils témoignent du lien physiopathologique reconnu entre certains variants du gène ABCB4 et le risque de développer un cancer hépato-biliaire.11
Prise en charge médicale en première intention
L’acide ursodésoxycholique (AUDC) constitue le traitement médical de choix du syndrome LPAC. L’AUDC est un acide biliaire hydrophile, inducteur du transporteur biliaire MDR3, qui permet, d’une part, de stimuler la sécrétion des phospholipides dans la bile et, d’autre part, de protéger l’épithélium biliaire contre l’effet toxique des acides biliaires endogènes. Même si le niveau de preuve de son efficacité dans le syndrome LPAC est faible en raison de l’absence d’étude contrôlée, le service médical rendu en pratique clinique est important et les effets bénéfiques sur les symptômes sont souvent spectaculaires. De ce fait, l’AUDC est reconnu actuellement comme le seul traitement de référence du syndrome LPAC, administré à la posologie initiale de 5 à 10 mg/kg/j. En cas de symptômes persistants, cette posologie peut être augmentée à 15 mg/kg/j.12 L’efficacité de l’AUDC n’est pas toujours immédiate et doit être évaluée après au moins trois mois de traitement. La prise d’AUDC est le plus souvent associée à une diminution en fréquence et en intensité des crises de colique hépatique et des complications biliaires. Toutefois, jusqu’à 20 % des patients pourraient être en échec, partiel ou complet, du traitement par AUDC.13 La réponse clinique à l’AUDC est parfois dissociée de la réponse radiologique. En conséquence, la persistance de signes échographiques de microlithiase intrahépatique ne doit pas justifier à elle seule l’intensification du traitement. L’objectif thérapeutique est essentiellement clinique, visant prioritairement à contrôler les symptômes biliaires.
En cas de réponse clinique insuffisante à l’AUDC, aucun traitement de deuxième intention ne fait actuellement l’objet d’un consensus ni d’un niveau de preuve suffisant. Toutefois, on peut proposer un traitement complémentaire par ézétimibe à la posologie de 10 mg/j, qui a pour effet de diminuer la concentration du cholestérol dans la bile et le risque de lithiase biliaire chez l’animal, ou éventuellement un traitement inducteur du transporteur MDR3, tel que les fibrates ou l’acide obéticholique.14 - 16 Le recours à ce type de traitement doit être discuté et validé dans un centre expert, après avoir vérifié l’observance du traitement par AUDC et éliminé certains diagnostics différentiels, comme la dysfonction du sphincter d’Oddi ou une douleur abdominale d’autre origine (trouble fonctionnel intestinal).
Indications du traitement endoscopique
L’extraction des calculs biliaires par voie endoscopique rétrograde (cholangio-pancréatographie rétrograde par voie endoscopique, CPRE) peut être nécessaire en cas de lithiase biliaire symptomatique (avec ou sans complication) de la voie biliaire principale ou des canaux hépatiques principaux. Dans une série rétrospective de patients atteints de syndrome LPAC, le recours à la CPRE avait été nnécessaire pour 30 % d’entre eux, le plus souvent avant que le traitement par AUDC ait été débuté.13 Après l’instauration du traitement par AUDC, seule une minorité de patients a nécessité un traitement endoscopique. Les indications de CPRE rejoignaient celles de la lithiase biliaire commune, à savoir la lithiase symptomatique de la voie biliaire principale (54 %) et les angiocholites lithiasiques (30 %). Les calculs étaient le plus souvent cholestéroliques, c’est-à-dire d’aspect jaunâtre et mou. Ils peuvent aussi être en partie pigmentaires (bruns), notamment chez les patients ayant présenté des épisodes répétés d’angiocholite. Les résultats de la CPRE étaient bons, avec un taux de succès supérieur à 80 %. Le risque de pancréatite aigüe post-CPRE était identique dans la population LPAC (7 %) par rapport à la population générale.
Prise en charge chirurgicale
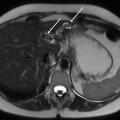
La cholécystectomie doit être évitée dans la mesure du possible car elle ne prévient pas le risque de récidive d’événement biliaire, qui est lié, dans la très grande majorité des cas, à la lithiase intrahépatique et non à la lithiase vésiculaire. La lithiase vésiculaire n’est présente que dans 30 % des cas de syndrome LPAC. En revanche, en cas de cholécystite aiguë lithiasique avérée, la cholécystectomie doit bien sûr être réalisée afin de limiter le risque de complications (perforation, abcès, péritonite). Une hépatectomie partielle peut être proposée en cas d’empierrement des voies biliaires intrahépatiques, symptomatique ou compliqué (angiocholites, abcès) et en cas d’échec du traitement par AUDC, si cet empierrement est limité à un lobe ou à un segment du foie (fig. 3).
La transplantation hépatique peut être l’ultime recours en cas d’empierrement diffus des voies biliaires intrahépatiques compliqué d’angiocholites à répétition ou de cirrhose biliaire secondaire mettant en jeu le pronostic vital.
Mesures associées
L’arrêt des contraceptifs oraux ne doit être proposé qu’en cas d’échec du traitement par AUDC.
En situation d’obésité ou de surpoids, la réduction pondérale doit faire partie de la prise en charge globale de la maladie.
Place du dépistage familial
Il n’existe pas de recommandation concernant le dépistage des apparentés des patients atteints du syndrome LPAC. Comme indiqué précédemment, la majorité des cas index diagnostiqués en pratique clinique sont sporadiques. Par ailleurs, il n’existe pas de consensus sur l’intérêt et l’efficacité de la prise d’AUDC au long cours chez les patients asymptomatiques ne présentant que des signes échographiques de la maladie (forme infraclinique) et/ou porteurs d’un variant pathogène du gène ABCB4. En pratique, il ne semble pas déraisonnable de proposer une échographie hépatique de dépistage chez les frères et sœurs, voire les enfants à leur majorité, des sujets atteints, en particulier quand ces derniers n’ont pas de variant pathogène identifié du gène ABCB4. En revanche, en cas de variant identifié, le dépistage génétique doit être privilégié et suivi, en cas de positivité, par une échographie hépatique. La prescription d’AUDC est discutée au cas par cas, en fonction des facteurs de risque de complication (symptômes, macrolithiase, grossesse, obésité) et des souhaits du sujet dépisté.
Surveillance clinique
La surveillance du syndrome LPAC repose sur un examen clinique, initialement semestriel, permettant d’évaluer la tolérance et la réponse au traitement par AUDC. Une surveillance des tests sanguins hépatiques doit être réalisée en cas d’anomalies biologiques initiales. En cas de réponse clinique et biologique favorable ou satisfaisante, la surveillance peut être élargie (une consultation tous les un à deux ans).
La place de l’échographie hépatique dans le suivi du syndrome LPAC reste à définir en raison de la discordance possible entre les réponses clinique et radiologique. Toutefois, il est souhaitable de faire une échographie de contrôle après six à douze mois de traitement par l’AUDC et/ou en cas de réapparition de symptômes de type biliaire.
Les récentes recommandations de l’Association française pour l’étude du foie (AFEF) sur la prise en charge du cholangiocarcinome ne proposent pas de dépistage radiologique systématique du cholangiocarcinome chez les patients atteints de syndrome LPAC.17 Sur avis d’experts, cette recommandation peut être nuancée, en particulier chez les patients ayant une forme macrolithiasique et cholangiopathique de la maladie, à plus haut risque de transformation maligne, chez lesquels une surveillance radiologique annuelle peut être proposée, en alternant IRM et échographie.
Forme rare de lithiase biliaire intrahépatique
Le syndrome LPAC est une forme rare de lithiase biliaire intrahépatique d’origine génétique atteignant typiquement de jeunes adultes de moins 40 ans, trois fois plus souvent des femmes que des hommes. Le syndrome LPAC est historiquement associé à des anomalies génétiques du transporteur des phospholipides MDR3 (ABCB4), mais un variant pathogène n’est retrouvé que chez 30 à 50 % des patients, indiquant une hétérogénéité génétique de la maladie. Si un traitement endoscopique est rapporté chez près d’un tiers des patients avant le diagnostic, le traitement de référence est l’administration orale d’AUDC au long cours, qui a un effet bénéfique symptomatique pour une majorité de patients.
Caractéristiques phénotypiques particulières du syndrome LPAC associé à une mutation d’ABCB4
Augmentation plus fréquente des gamma GT (cholestase chronique)
Augmentation de la fréquence de formes plus sévères de LPAC définies par :
- calculs vrais intrahépatiques avec dilatation des voies biliaires ;
- lithiase de la voie biliaire principale
Fréquence plus élevée de cholangiocarcinome (sujet index et apparentés)
2. Stättermayer AF, Halilbasic E, Wrba F, et al. Variants in ABCB4 (MDR3) across the spectrum of cholestatic liver diseases in adults. J Hepatol 2020;73(3):651‑63.
3. Poupon R, Rosmorduc O, Boëlle PY, et al. Genotype-phenotype relationships in the low-phospholipid-associated cholelithiasis syndrome: A study of 156 consecutive patients. Hepatology 2013;58(3):1105‑10.
4. Dong C, Condat B, Picon-Coste M, et al. Low-phospholipid-associated cholelithiasis syndrome: Prevalence, clinical features, and comorbidities. JHEP Rep 2021;3(2):100201.
5. Delaunay JL, Durand-Schneider AM, Dossier C, et al. A functional classification of ABCB4 variations causing progressive familial intrahepatic cholestasis type 3. Hepatology 2016;63(5):1620‑31.
6. Condat B, Zanditenas D, Barbu V, et al. Prevalence of low phospholipid-associated cholelithiasis in young female patients. Dig Liver Dis Off J Ital Soc Gastroenterol Ital Assoc Study Liver 2013;45(11):915‑9.
7. Gouveia C, Flor de Lima M, Pereira F, et al. Should patients with symptomatic cholelithiasis before 30 years of age be tested for ABCB4 gene mutations? Scand J Gastroenterol 2020;55(8):958‑62.
8. Avena A, Puggelli S, Morris M, et al. ABCB4 variants in adult patients with cholestatic disease are frequent and underdiagnosed. Dig Liver Dis 2021;53(3):329‑44.
9. Su S, Trinh A, Metz AJ, et al. Targeted liver ultrasound performed by an expert is the pivotal imaging examination for low phospholipid-associated cholelithiasis. Eur J Gastroenterol Hepatol 2023;35(3):327‑32.
10. Biyoukar M, Corpechot C, El Mouhadi S, et al. ABCB4 variant is associated with hepatobiliary MR abnormalities in people with low-phospholipid-associated cholelithiasis syndrome. JHEP Rep 2022;4(11):100590.
11. Gudbjartsson DF, Helgason H, Gudjonsson SA, et al. Large-scale whole-genome sequencing of the Icelandic population. Nat Genet 2015;47(5):435‑44.
12. Rosmorduc O, Poupon R. Low phospholipid associated cholelithiasis: Association with mutation in the MDR3/ABCB4 gene. Orphanet J Rare Dis 2007;2:29.
13. Salin G, Corpechot C, Ouazana S, et al. Endoscopic features of low-phospholipid-associated cholelithiasis syndrome: A retrospective cohort study. Clin Res Hepatol Gastroenterol 2024;48(5):102324.
14. Wang HH, Portincasa P, Mendez-Sanchez N, et al. Effect of ezetimibe on the prevention and dissolution of cholesterol gallstones. Gastroenterology 2008;134(7):2101‑10.
15. Nakamuta M, Fujino T, Yada R, et al. Therapeutic effect of bezafibrate against biliary damage: A study of phospholipid secretion via the PPARalpha-MDR3 pathway. Int J Clin Pharmacol Ther 2010;48(1):22‑8.
16. Soret PA, Lemoinne S, Mallet M, et al. Obeticholic acid as a second-line treatment for low phospholipid-associated cholelithiasis syndrome. Aliment Pharmacol Ther 2024;59(1):113‑7.
17. Recommandations AFEF sur la prise en charge des cholangiocarcinomes intrahépatiques et périhilaires. Novembre 2022. https://urls.fr/XrJLGs.
Dans cet article
- Physiopathologie du syndrome LPAC
- Caractérisation génétique
- Données épidémiologiques
- Présentations clinique et radiologique
- Diagnostic positif
- Complications lithiasiques du syndrome LPAC
- Prise en charge médicale en première intention
- Indications du traitement endoscopique
- Prise en charge chirurgicale
- Mesures associées
- Place du dépistage familial
- Surveillance clinique
- Forme rare de lithiase biliaire intrahépatique
 Encadrés
Encadrés