Connaître les médicaments susceptibles d’induire un syndrome parkinsonien.
Décrire les principes de la prise en charge.
Épidémiologie (rang C)
La maladie débute en général dans la sixième décennie mais peut survenir à tout âge (10 % avant 40 ans).
La MP est la deuxième cause de handicap moteur d’origine neurologique chez le sujet âgé (après les accidents vasculaires cérébraux).
Le diagnostic de certitude de MP est histologique et ne peut pas se faire du vivant du patient. Le diagnostic actuel repose donc sur des données cliniques ; différentes techniques, en particulier d’imagerie fonctionnelle, commencent à améliorer sa fiabilité et sa probabilité.
Physiopathologie (rang B)
Le processus neurodégénératif dépasse la voie nigrostriée, expliquant la survenue d’autres signes moteurs (signes axiaux, par exemple) et non moteurs (troubles cognitifs, par exemple) résistant au traitement dopaminergique.
Les lésions pathologiques évoluent en aspect et en sévérité de façon continue et avec une séquence de modifications prévisibles en six stades neuropathologiques :
- stades présymptomatiques 1 et 2 qui pourraient durer jusqu’à dix ans ; les lésions concernent le tractus olfactif, le noyau dorsal du nerf vague, les neurones du système digestif et le locus cæruleus ;
- stades 3 et 4 ; les signes moteurs apparaissent lorsque la perte progressive des neurones dopaminergiques de la pars compacta du locus niger à l’origine de la voie nigrostriée atteint 60 % (premier stade symptomatique). Le mésocortex et d’autres noyaux sous-corticaux (nucleus basalis de Meynert) dégénèrent également ;
- stades 5 et 6 ; après de nombreuses années, le néocortex primaire, secondaire et associatif dégénère, occasionnant des troubles moteurs, cognitifs et comportementaux résistants au traitement dopaminergique.
Les facteurs étiologiques principaux sont au nombre de cinq :
- environnementaux, après la découverte d’un syndrome parkinsonien chez des toxicomanes après injection d’un produit chimique, le 1-méthyl-4-phényl-1,2,3,6-tétrahydropyridine ( MPTP ), toxique pour les neurones dopaminergiques. Les insecticides (roténone) et les herbicides (Paraquat) ont une structure chimique proche du MPTP. La prévalence de la maladie est plus élevée dans les régions industrialisées (chimie, pesticides) et dans les zones rurales (usage intensif d’insecticides). Si une origine toxique exogène constitue un facteur causal potentiel, il est probable qu’il existe une susceptibilité individuelle génétique à ces toxiques ;
- une relation inverse entre la consommation de tabac et la survenue d’une MP semble bien établie ; elle pourrait être liée à un éventuel rôle neuroprotecteur du tabac ou serait le témoin d’une personnalité prémorbide ;
- la consommation de café semble également réduire le risque de MP ;
- l’existence d’un facteur génétique est corroborée par le fait que 15 à 20 % des patients rapportent l’atteinte d’un autre membre de la famille (le risque relatif des apparentés au premier degré de patient est 3 à 4 fois supérieur à celui des apparentés de témoins) ;
- dans la majorité des cas, la MP est sporadique et vraisemblablement d’origine multifactorielle, avec l’implication de facteurs génétiques et environnementaux.
Manifestations cliniques
Symptomatologie initiale
Tremblement de repos
Dans 70 % des cas, il s’agit d’un signe initial.Dans 5 à 10 % des cas, le tremblement est d’abord postural. Décrit comme une sensation de vibration interne, puis visible, il se majore ou apparaît à l’émotion et à l’épreuve du calcul mental. Il est unilatéral ou très asymétrique. Il débute classiquement au niveau des membres supérieurs, mais intéresse parfois de façon isolée le pied. Il peut aussi concerner les lèvres, la mâchoire, et exceptionnellement le chef.
Présent au repos, il disparaît lors du mouvement. Il est lent (fréquence 4-6 Hz). Parfois, il est absent.
Syndrome akinéto-hypertonique
Présent dans 20 à 30 % des cas et plus insidieux, il peut être diagnostiqué avec retard.Il se traduit, selon les cas, par une perte du balancement d’un bras à la marche, une gêne segmentaire limitée au niveau d’un membre supérieur (gestes répétitifs tels que se raser, battre des œufs, lenteur gestuelle, gêne à l’écriture avec micrographie progressive) ou d’un membre inférieur, parfois par une simple fatigabilité ou raideur à la marche avec l’impression d’un pied qui traîne, enfin par une micrographie isolée.
De topographie initialement unilatérale, il peut être bilatéral d’emblée mais asymétrique.
Troubles de la marche et de la posture
Ils sont rarement constatés au stade initial de la maladie, sauf lorsque l’akinésie prédomine au niveau des membres inférieurs. Ils sont plus fréquents et plus précoces chez les sujets âgés.Leur apparition isolée doit faire rechercher une autre cause que la maladie de Parkinson (voir le paragraphe sur les diagnostics différentiels), par exemple un syndrome parkinsonien vasculaire.
Chez le sujet de moins de 40 ans
La maladie débute volontiers par une dystonie focalisée : dystonie du pied en varus équin ou en extension du gros orteil à la course à pied. Il existe alors souvent un discret syndrome akinéto-rigide associé.Formes de début trompeuses
Elles sont le plus souvent associées à un syndrome akinéto-rigide. Un syndrome dépressif isolé avec ou sans apathie sans facteur déclenchant peut révéler la maladie, de même que des douleurs de l’épaule associées à une raideur, un déficit de l’odorat par atteinte du noyau du nerf olfactif, ou des troubles du comportement en sommeil paradoxal (rêves animés, parfois responsables d’actes auto- ou hétéro-agressifs).Ces symptômes peuvent précéder les troubles moteurs de plusieurs années au cours de la phase prodromale.
Diagnostic
Établir le diagnostic d’un syndrome parkinsonien
Il se définit par la présence d’une akinésie associée à au moins l'un des symptômes suivants : tremblement de repos, rigidité extrapyramidale ou instabilité posturale.Le tremblement prédomine au niveau de l’hémicorps où il a débuté ; dans les formes sévères, le maintien d’attitude et le mouvement volontaire ne suppriment pas totalement le tremblement et peuvent parfois en augmenter la fréquence.
Des signes d’akinésie (ralentissement à l’initiation), de bradykinésie (ralentissement à l’exécution des mouvements) ou d’hypokinésie (réduction d’amplitude des mouvements) se caractérisent par un faciès inexpressif et figé avec une hypomimie et une rareté du clignement palpébral, un ralentissement lors de la réalisation des gestes alternatifs rapides (opposition pouce-index, fermeture-ouverture de la main, marionnette), une perte du ballant du bras lors de la marche, une micrographie, une gêne aux mouvements alternatifs (battre la mesure) au niveau des membres inférieurs, une démarche lente, une élocution moins bien articulée, monocorde et monotone avec parfois accélération paradoxale du débit verbal (tachyphémie). Tous les mouvements du quotidien peuvent être concernés.
La rigidité plastique (sensibilisation par la manœuvre de Froment) cède en tuyau de plomb ou par à-coups (phénomène de « roue dentée »), prédomine sur les muscles fléchisseurs, se majore à la fatigue, au froid et s’atténue au cours du sommeil. Elle rend compte des déformations posturales et des douleurs et joue un rôle dans l’attitude en flexion des segments de membres, du cou et du tronc.
Les troubles de la marche sont modérés au début de la maladie. Progressivement, la marche se fait à petits pas. Les difficultés sont majorées au démarrage (retard d’initiation à la marche), au demi-tour ou lors de franchissement d’obstacles ou du passage d’une porte.
À un stade plus tardif, le malade a tendance à courir après son centre de gravité en accélérant le pas (festination) ; les enrayages cinétiques (blocages ou freezing) peuvent durer plusieurs secondes et céder lorsque l’on demande au patient d’enjamber un obstacle. L’instabilité posturale apparaît à un stade évolué de la maladie, entraînant des chutes en rétropulsion.
Exclure une cause iatrogène
Il faut rechercher systématiquement la prise de traitements susceptibles de déclencher un syndrome parkinsonien, tels que des neuroleptiques (parfois cachés). Il est essentiel de consulter les résumés des caractéristiques des produits (RCP) au moindre doute.Vérifier l’absence de critères d’exclusion de la maladie de Parkinson
Il faut rechercher systématiquement des signes cliniques dont l’association précoce au syndrome parkinsonien va à l’encontre du diagnostic de maladie de Parkinson ; ce sont les drapeaux rouges : chutes précoces, signes précoces d’atteinte cognitive, pseudobulbaire (dysarthrie et dysphagie) ou de dysautonomie (incontinence urinaire, hypotension orthostatique sévère, impuissance), syndrome cérébelleux, atteinte pyramidale, troubles oculomoteurs et signes corticaux (apraxie, aphasie, astéréognosie, myoclonies), puis, avec le temps, absence de réponse prolongée au traitement dopaminergique (au-delà de cinq ans), progression rapide de la maladie avec handicap sévère.Soulignons que certains de ces symptômes peuvent cependant être présents à un stade plus tardif de la maladie (par exemple les troubles sphinctériens, l’instabilité posturale et la démence).
Réponse à la L-dopa
Une franche amélioration de la symptomatologie lors de la mise en route du traitement dopaminergique constitue également un critère diagnostique essentiel (la dopasensibilité dure plus de cinq ans) avec l’apparition dans un second temps de dyskinésies.Signes associés non moteurs (rang B)
Signes neurovégétatifs
Hypersialorrhée précoce et hyperséborrhée donnent un aspect de visage pommadé.Les troubles digestifs possibles sont les suivants : constipation, motricités gastrique et œsophagienne ralenties entraînant une symptomatologie à type de hernie hiatale avec hoquet. L’hypotension artérielle orthostatique est souvent d’apparition tardive.
Enfin, troubles vésico-sphinctériens (impériosités mictionnelles, incontinence tardive) et troubles vaso-moteurs avec froideur des extrémités sont à rechercher.
Troubles sensitifs
Ils sont très variables d’un sujet à l’autre : crampes, engourdissement, picotements, sensation de chaleur ou de froid au niveau des extrémités, localisés du côté où la symptomatologie extrapyramidale prédomine.Les principales localisations sont les chevilles, les poignets, les épaules, le rachis cervical et lombaire.
Troubles du sommeil et de la vigilance
Le sommeil peut être perturbé par les troubles sphinctériens (pollakiurie), des difficultés motrices nocturnes (renforcement du syndrome parkinsonien) et des troubles du comportement en sommeil paradoxal (cauchemars).L’insomnie est initiale ou survient en deuxième partie de nuit.
La somnolence diurne est parfois favorisée par les traitements, surtout les agonistes dopaminergiques.
Troubles cognitifs et du comportement
Voir paragraphe dédié plus bas.Principales phases évolutives
L’instauration d’un traitement substitutif permet un contrôle satisfaisant des symptômes pendant au moins cinq années (en tenant compte de la tolérance aux différents traitements). Au cours de cette première étape, la maladie est susceptible de s’aggraver au décours de phénomènes intercurrents (infection, par exemple).
La deuxième période est celle des complications motrices liées aux traitements et à l’évolution du processus dégénératif : fluctuations motrices et non motrices et dyskinésies induites par la lévodopa.
Puis on constate progressivement l’émergence de signes moteurs axiaux tardifs. C’est à ce stade que les troubles cognitifs et comportementaux sont susceptibles d’être observés. On parle de déclin moteur et cognitif.
Au dernier stade, la marche peut devenir impossible (phénomène inconstant : tous les patients ne terminent pas leur vie en fauteuil roulant) ; le patient perd son autonomie et doit être aidé pour les différentes activités de la vie quotidienne.
Investigations paracliniques (rang C)
Une imagerie fonctionnelle (scintigraphie au DAT-scan ou TEP-scan à la fluorodopa) est indiquée en cas de doute diagnostique entre un tremblement parkinsonien et un tremblement essentiel. L’indication est posée par le neurologue. Cet examen n’est justifié ni devant un syndrome parkinsonien typique ni pour évaluer le suivi. Il détecte une perte de terminaisons neuronales dopaminergiques fonctionnelles dans le striatum chez les patients ayant un syndrome parkinsonien cliniquement douteux. Il est anormal dans la maladie de Parkinson et également dans d’autres syndromes parkinsoniens dégénératifs. Il est normal en cas de tremblement essentiel, de syndrome parkinsonien d’origine médicamenteuse et de tremblement dystonique.
L'imagerie par résonance magnétique (IRM) est utilisée chez les patients pour qui il est cliniquement utile d’identifier le degré de maladie cérébrovasculaire (diagnostic différentiel entre MP et syndrome parkinsonien vasculaire), le degré et la localisation de l’atrophie corticale (suspicion d’un autre syndrome parkinsonien dégénératif).
Il n’est pas recommandé d’utiliser la tomodensitométrie ni l’IRM dans le diagnostic de routine de la maladie de Parkinson, de même que l’échographie transcrânienne ou les tests olfactifs.
Complications motrices liées au traitement dopaminergique (rang B)
Fluctuations d’efficacité
Plus de 50 % des patients présentent des fluctuations en moyenne après cinq ans de traitement par L-dopa ; elles sont finalement constantes dans l’évolution.
Elles s’expliquent essentiellement par le fait que les médicaments dopaminergiques ont une demi-vie plasmatique brève et qu’avec l’évolution, le cerveau perd progressivement ses capacités de stockage de la dopamine.
Ce sont surtout les signes moteurs qui se majorent au cours de ces fluctuations :
- akinésie de fin de dose avec un raccourcissement progressif de l’effet de chaque prise de L-dopa, la symptomatologie extrapyramidale réapparaissant avant la prise suivante ;
- akinésie matinale, akinésie nocturne, akinésie nycthémérale survenant à horaires réguliers, souvent l’après-midi ;
- puis la réapparition de la symptomatologie parkinsonienne est plus anarchique (phénomène « on-off ») avec passages assez brutaux d’un état non parkinsonien à un état parkinsonien sévère.
Mouvements involontaires, ou dyskinésies
Ils sont préférentiellement observés chez des patients débutant la maladie à un âge relativement précoce (moins de 60 ans), surtout si des doses fortes de L-dopa ont été prescrites. L’évolution de la dénervation favorise avec le temps l’apparition de ces dyskinésies.
On distingue :
- les dyskinésies de milieu de dose contemporaines de taux sériques élevés de L-dopa (stimulation dopaminergique excessive) qui se caractérisent par des mouvements choréiques ou choréo-athétosiques des membres, du tronc ou de la région cervicale ;
- les dyskinésies biphasiques : en début de dose, elles annoncent l’efficacité thérapeutique et sont volontiers caractérisées par des mouvements balliques ou des mouvements alternatifs répétitifs des membres inférieurs ; en fin de dose, elles annoncent le retour à l’état parkinsonien, il s’agit alors de postures dystoniques des membres inférieurs douloureuses ;
- des mouvements dystoniques (pied en varus équin, extension spontanée du gros orteil) sont également observés au cours des périodes de blocage ou le matin au réveil avant la première prise médicamenteuse.
Signes tardifs (rang B)
Signes moteurs axiaux
Progressivement, l’attitude générale du patient est en flexion. Les déformations du rachis accentuent également l’altération du contrôle postural avec, dans certains cas, une camptocormie (forme extrême d’antéflexion réductible du tronc).
Détérioration cognitive
Troubles psychiques
Le traitement dopaminergique peut provoquer des hallucinations élémentaires parfois critiquées (hallucinoses, par exemple, avec impressions de passage ou de présence) mais aussi de véritables délires correspondant à une psychose dopaminergique (de type hallucinatoire ou paranoïaque). La survenue de ces complications peut être l’indicateur d’une évolution de la maladie vers un état démentiel associé.
Le risque de survenue de ces troubles psychiques augmente avec l’âge, la sévérité des troubles moteurs, des signes dépressifs et des troubles cognitifs.
Diagnostic différentiel
Ce qui n’est pas un syndrome parkinsonien
Chez le sujet âgé, les troubles de la marche à petits pas peuvent être consécutifs à une hydrocéphalie chronique (importante instabilité posturale, tendance à la rétropulsion, élargissement du polygone de sustentation, troubles sphinctériens, détérioration intellectuelle) mais aussi à des lésions ischémiques multiples des noyaux gris centraux.
Autres syndromes parkinsoniens
Syndrome parkinsonien iatrogène
Une cause iatrogène doit être recherchée systématiquement devant tout syndrome parkinsonien.Le syndrome iatrogène est principalement induit par les neuroleptiques (antipsychotiques) ou neuroleptiques « cachés » (antinauséeux comme le métoclopramide et la métopimazine, sédatif comme l’alimémazine), qu’il s’agit de rechercher méticuleusement à l’interrogatoire.
Des inhibiteurs calciques de type flunarizine (traitement de fond de la migraine) et des antidépresseurs sont plus rarement responsables d’un syndrome parkinsonien.
Au moindre doute, il faut consulter les résumés des caractéristiques des produits (RCP).
Cliniquement, le syndrome parkinsonien est plutôt symétrique (caractéristique inconstante), plus fréquemment un tremblement postural ou d’action qu’un authentique tremblement de repos, avec présence potentielle de dyskinésies bucco-linguo-faciales, et absence de réponse au traitement dopaminergique.
Autres syndromes parkinsoniens dégénératifs (rang C)
Ils se distinguent de la MP par la faible ou l’absence de réactivité au traitement dopaminergique résultant de lésions post-synaptiques et par l’existence de signes neurologiques associés.L’atrophie multisystématisée comporte un syndrome parkinsonien peu dopasensible s’accompagnant de signes axiaux, d’une dysarthrie et de troubles posturaux précoces, un syndrome cérébelleux (essentiellement statique), un syndrome dysautonomique précoce et constant au cours de l’évolution (hypotension orthostatique, troubles génito-sphinctériens, troubles vasomoteurs), un syndrome pyramidal.
La paralysie supranucléaire progressive se caractérise par un syndrome parkinsonien symétrique à prédominance axiale, doparésistant avec troubles posturaux précoces (chutes en rétropulsion), une paralysie supranucléaire de l’oculomotricité verticale, un syndrome pseudobulbaire et une démence précoce.
Le syndrome de dégénérescence corticobasale est beaucoup plus rare, associant un syndrome parkinsonien très asymétrique, avec dystonie, apraxie, syndrome pyramidal et syndrome frontal.
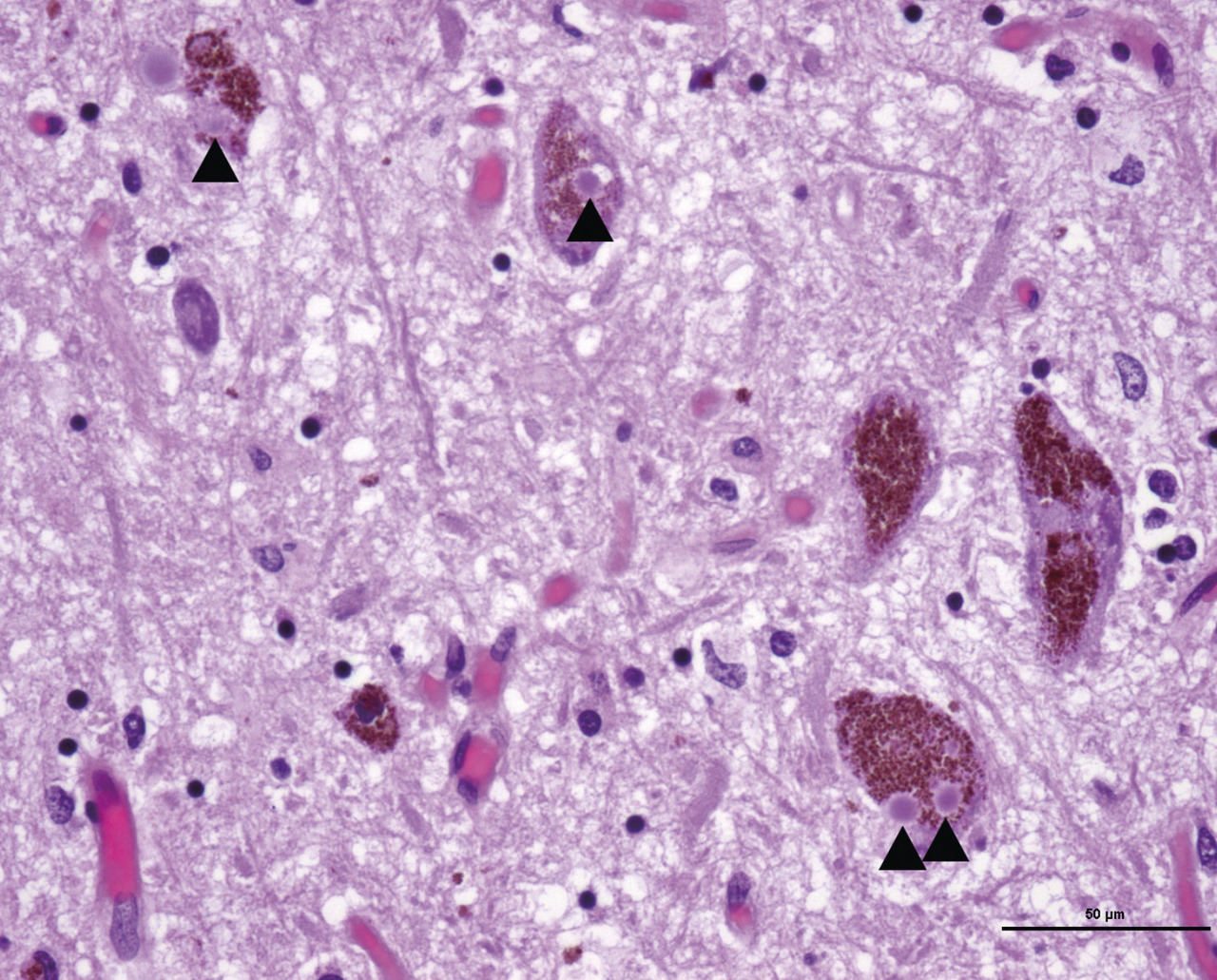
Dans la démence à corps de Lewy (
Maladie de Wilson (rang C)
C’est une maladie familiale autosomique récessive responsable d’une accumulation de cuivre à l’origine de lésions du système nerveux et du foie, liée à un déficit d’excrétion par anomalie de transport du cuivre vers la bile associée à un déficit du transport plasmatique du cuivre (déficit de la céruléoplasmine). Le cuivre accumulé dans l’organisme a tendance à se concentrer dans le foie et dans le système nerveux central (les noyaux gris centraux : striatum, globus pallidus).Syndrome parkinsonien vasculaire (rang C)
Il est la conséquence de lésions vasculaires multiples touchant les noyaux gris centraux ou la substance blanche.L’IRM cérébrale permet de confirmer le diagnostic. Cliniquement, il se caractérise par un syndrome parkinsonien plutôt symétrique et peu sensible au traitement dopaminergique, la prédominance des troubles aux membres inférieurs (lower body parkinsonism) et une marche avec freezing précoce, un syndrome pseudobulbaire.
Prises en charge thérapeutiques (fig. 2 et 3)
Classes médicamenteuses (rang B)
Majoration des taux de dopamine : administration de son précurseur, la L-dopa, associé à un inhibiteur de la dopadécarboxylase périphérique
Au niveau cérébral, la L-dopa est transformée en dopamine par la dopadécarboxylase (DDC), présente aussi en dehors du système nerveux central. Il est adjoint à la L-dopa un inhibiteur de la DDC qui ne passe pas la barrière hémato-encéphalique dans les spécialités Modopar (lévodopa + benzérazide) et Sinemet (lévodopa + carbidopa).La dopathérapie est le traitement le plus efficace sur la symptomatologie parkinsonienne et le mieux toléré.
Stimulation directe des récepteurs dopaminergiques par les agonistes dopaminergiques
Il existe plusieurs spécialités (per os formes retard : Trivastal [piribédil], Requip [ropinirole], Sifrol [pramipexole], et en patch : Neupro [rotigotine]) et une forme injectable l’apomorphine [Apokinon stylo injectable]). Les formes orales ont une action moins puissante que la dopa et peuvent entraîner des effets indésirables dans 20 % des cas (addictions comportementales au jeu, hypersexualité, troubles des conduites alimentaires, conduites à risque nommées « troubles du contrôle des impulsions » mais aussi somnolence, œdèmes des membres inférieurs, prise de poids, hallucinations).Leur utilisation en début de maladie semble pouvoir retarder l’apparition des fluctuations motrices et des dyskinésies mais seulement à moyen terme (quelques années).
Inhibiteurs enzymatiques
Azilect (rasagiline) est un inhibiteur sélectif de la mono-amine-oxydase B (IMAO-B) qui aurait une action neuroprotectrice et peut être prescrit dès le début des symptômes.Des inhibiteurs de la catéchol-O-méthyl-transférase (ICOMT : Comtan [entacapone]) - qui réduisent la dégradation de L-dopa en périphérie - et Tasmar (tolcapone) - qui a une action périphérique et centrale - augmentent la biodisponiblité et la durée d’action de la L-dopa.
Stalevo (lévodopa + carbidopa + entacapone) associe un ICOMT à la L-dopa et à l’inhibiteur de la dopa-décarboxylase périphérique.
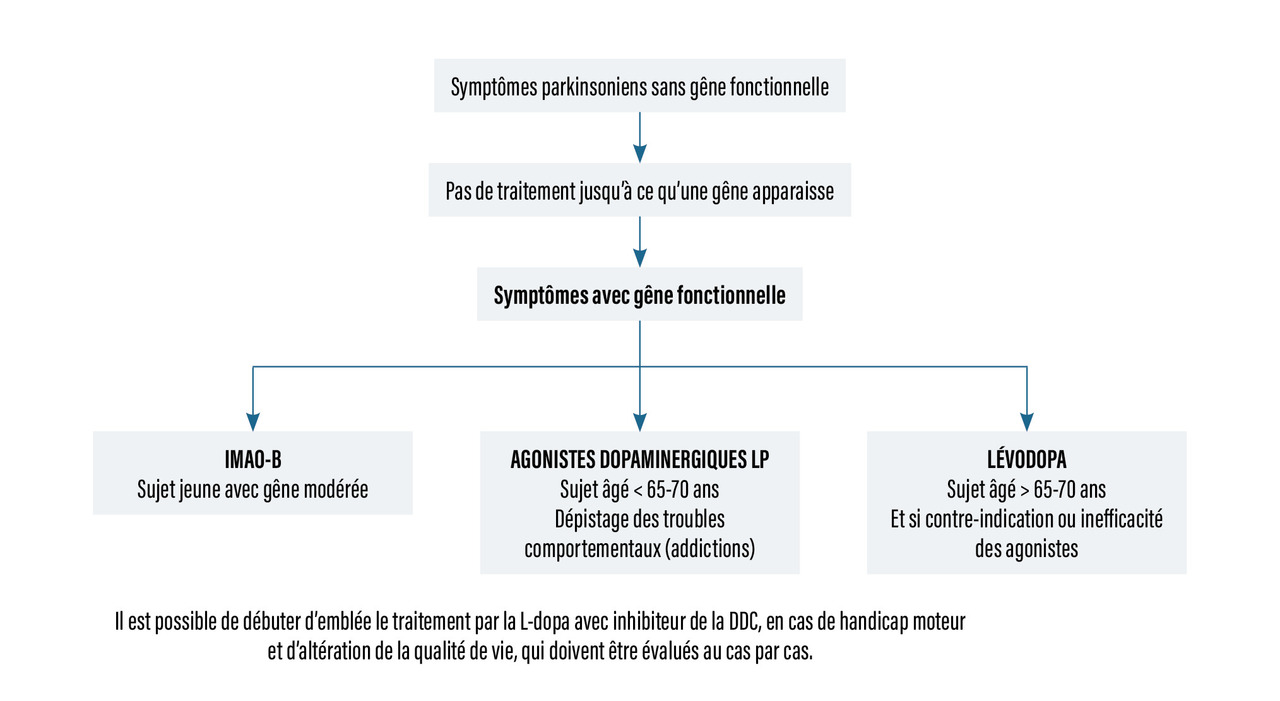
Principes de prescription (rang B)
Dans les formes débutant avant 65-70 ans, le traitement repose sur un agoniste dopaminergique LP seul, en monothérapie (retardement de l’apparition des complications motrices) ou un inhibiteur de la monoamine-oxydase B (IMAO-B). Ces deux thérapeutiques peuvent être associées. Cependant, en cas d’antécédent d’addiction, la prescription des agonistes dopaminergiques doit être évaluée avec beaucoup de prudence (risque potentiel de troubles du contrôle des impulsions). Dans l’idéal, un proche doit être présent pendant les consultations pour s’assurer de l’absence de ces troubles. Si le contrôle des symptômes n’est pas satisfaisant, il faut procéder à une progression posologique de l’agoniste dopaminergique.
Il est également possible de débuter d’emblée le traitement par la L-dopa avec inhibiteur de la DDC, surtout en cas de handicap moteur et d’altération de la qualité de vie qui doivent être évalués au cas par cas. La posologie doit être augmentée progressivement en trois ou quatre prises par jour (car demi-vie courte) avant les repas pour faciliter l’absorption et l’efficacité.
En cas de nausées ou de vomissements, un traitement par dompéridone peut être prescrit (diminution des effets indésirables liés à la stimulation des récepteurs dopaminergiques périphériques ; contre-indication en cas d’allongement de l'intervalle QTc) en limitant la prescription à la durée de traitement la plus courte (usuellement, 7 jours au maximum).
Dans les formes débutant après 65 à 70 ans, la L-dopa est prescrite seule (augmentation progressive jusqu’à la posologie minimale efficace) ou associée à un IMAO-C.
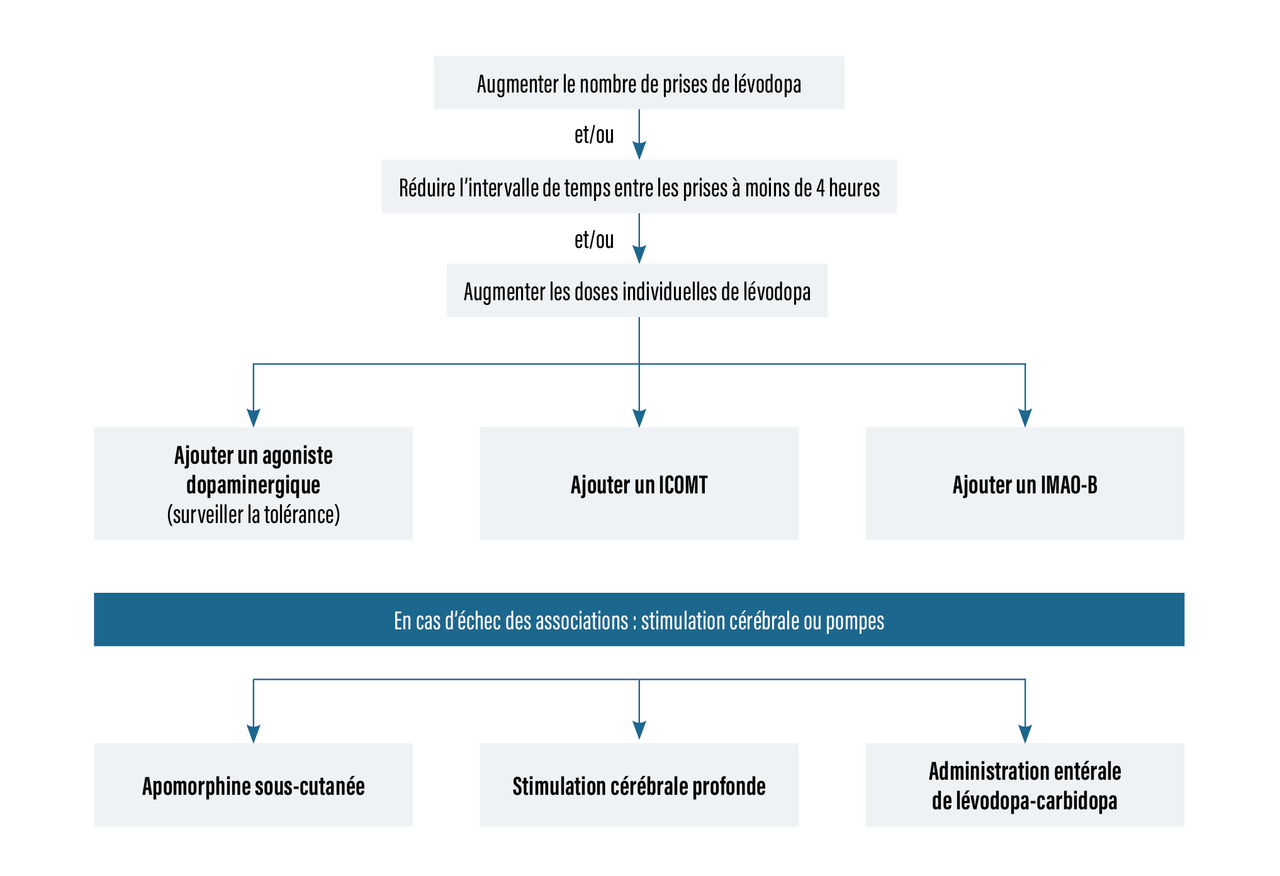
Prise en charge thérapeutique des stades plus avancés (fig. 3) [rang C]
Les dyskinésies peu invalidantes sont prises en charge par réduction des doses de L-dopa en cas de dyskinésies de milieu de dose avec fractionnement ; il est possible de prescrire de l’amantadine (Mantadix).
Pour les troubles psychiques de psychose parkinsonienne (confusion, hallucinations, délire), il est nécessaire d’arrêter les anticholinergiques, les agonistes dopaminergiques et les inhibiteurs enzymatiques et de maintenir une monothérapie par L-dopa à posologie réduite. Cette limitation du traitement, qui majore le handicap moteur, peut être évitée avec l’utilisation de la clozapine (Leponex), neuroleptique atypique, justifiant une surveillance régulière de l’hémogramme en raison des risques d’agranulocytose.
En cas de syndrome confusionnel, la première étape (comme face à tout syndrome confusionnel) est la recherche d’arguments cliniques et paracliniques en faveur d’une maladie générale (trouble métabolique, infection, hématome sous-dural…). La possibilité d’un facteur iatrogène doit aussi être évoquée : changements thérapeutiques récents.
La dépression, les troubles du sommeil et les troubles sphinctériens (constipation, mictions impérieuses) justifient une prise en charge spécifique. En cas de démence, un inhibiteur de l’acétylcholinestérase peut être prescrit, sous forme de patch : la rivastigmine (Exelon). Ce traitement, non remboursé, peut aussi limiter les hallucinations.
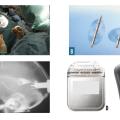
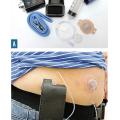
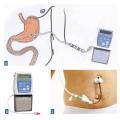
Traitements de recours : stimulation cérébrale et pompes (rang C)
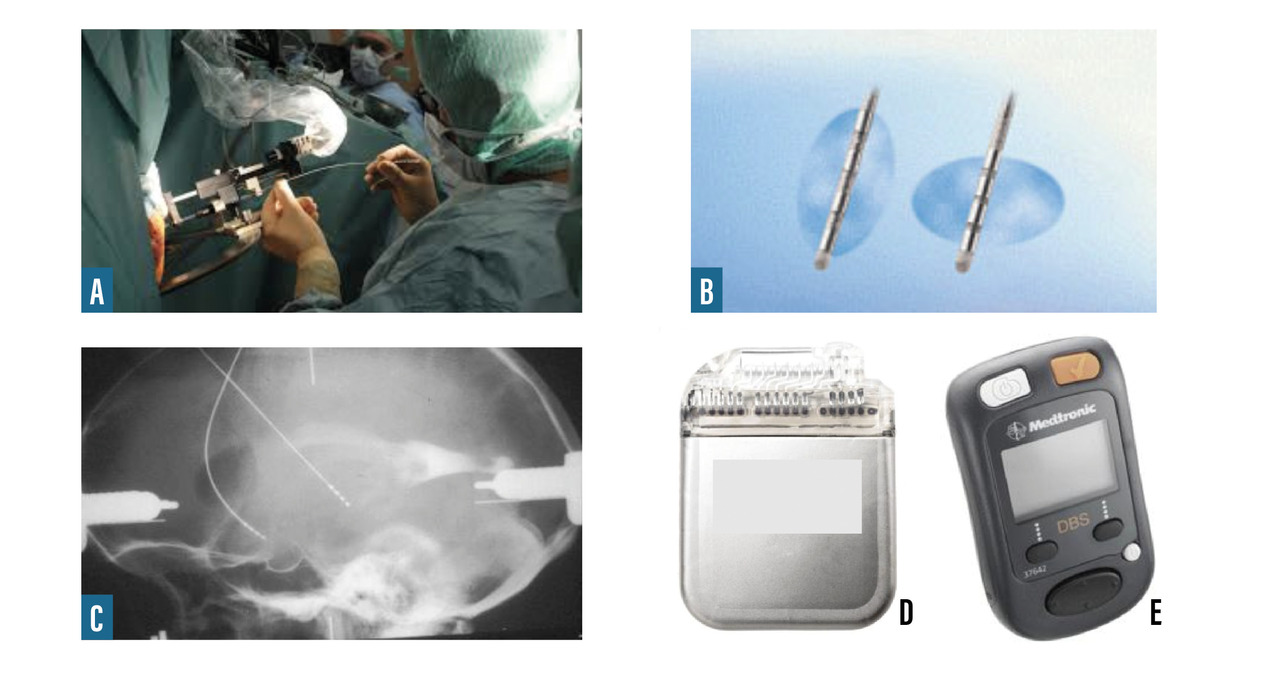
- la stimulation électrique chronique à haute fréquence (≥ à 130 Hz) des noyaux subthalamiques par des électrodes implantées par chirurgie stéréotaxique et reliées à un stimulateur placé en région pectorale peut être proposée chez des sujets relativement jeunes (moins de 70 ans), en l’absence de troubles sévères cognitifs ou du comportement (
fig. 4 ) ;
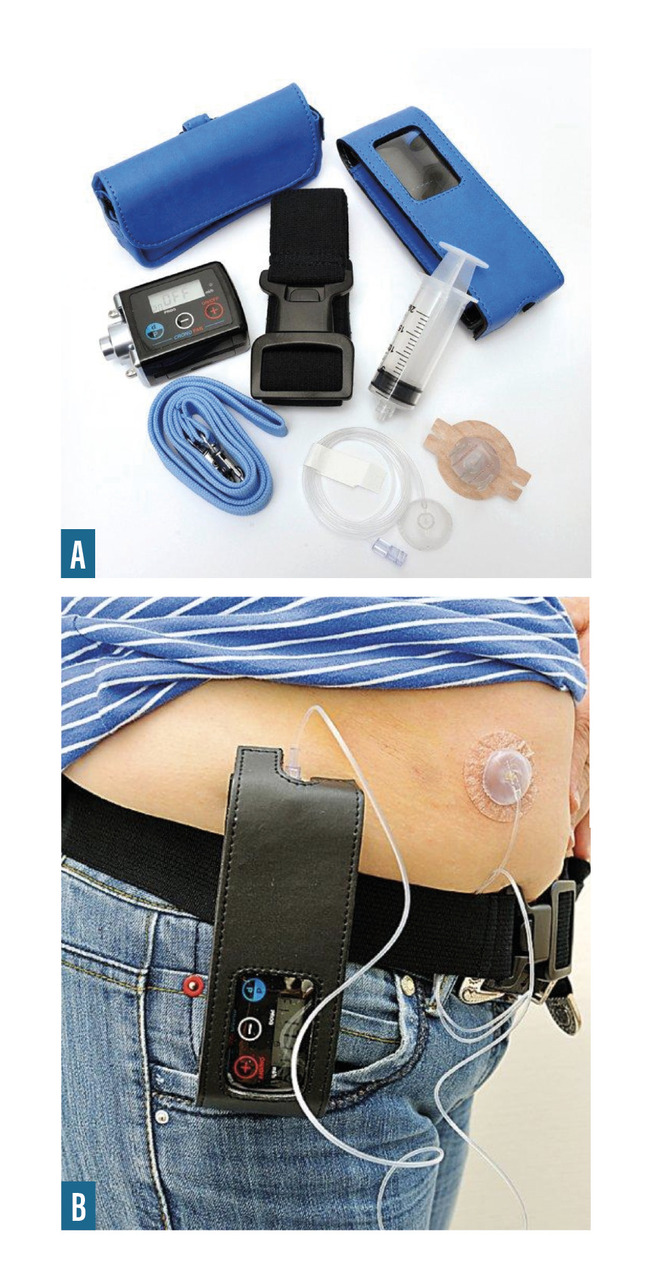
- pompe à apomorphine, pompe sous-cutanée délivrant pendant 12 ou 24 heures (à débits programmés différents si nécessaire) une dose continue de cet agoniste dopaminergique (
fig. 5 ) ;
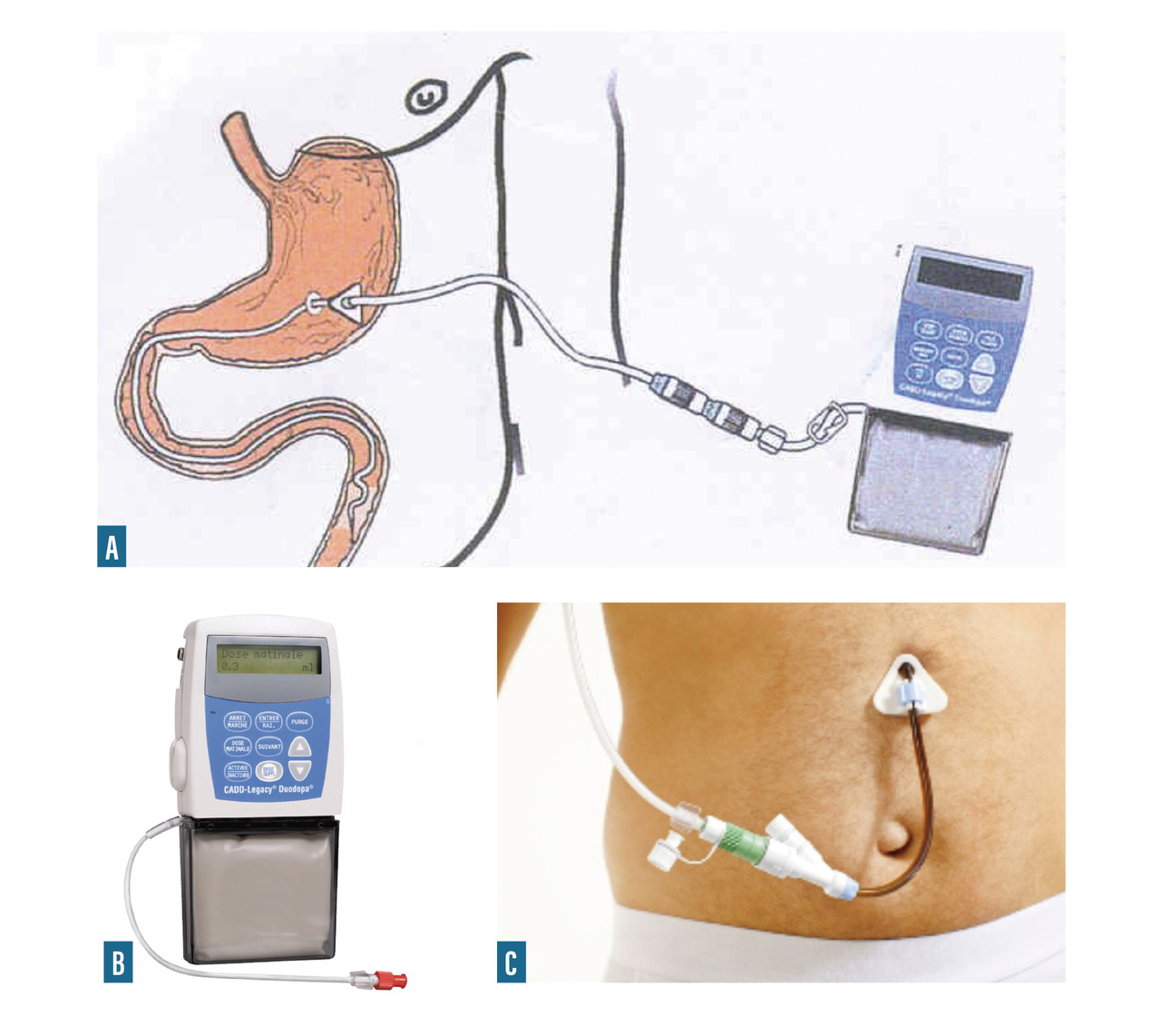
- Duodopa : infusion intra-duodéno-jéjunale d’un gel de L-dopa et inhibiteur de la dopadécarboxylase, après pose de gastrostomie (
fig. 6 ).
Autres mesures thérapeutiques
La prise en charge en kinésithérapie intensive au début de la maladie, puis centrée sur les signes axiaux (renforcement musculaire des membres inférieurs, lutte contre l’instabilité posturale et les enrayages cinétiques), est nécessaire tout au long de la maladie. Une activité physique et/ou sportive régulière est recommandée dès le début de la maladie.
La prise en charge des troubles de la parole et de la déglutition par une orthophoniste, selon la méthode LSVT (Lee Silverman Voice Treatment), est recommandée dès le début de la maladie.
Le suivi nutritionnel est adapté aux besoins du patient.
La prise en charge des troubles vésico-sphinctériens et dysautonomiques par des spécialistes concernés (urologues, médecins rééducateurs et cardiologues) annuelle est recommandée.
La demande de prise en charge à 100 % (affection de longue durée : ALD) doit être réalisée par le médecin traitant. Le renforcement des aides à domicile est effectué après évaluation des besoins par l’assistante sociale.
Un suivi neuropsychologique (dépistage des troubles cognitifs) et psychologique (thérapie cognitivo-comportementale, souvent à l’annonce du diagnostic ou dans le cours évolutif du patient et de l’aidant) est parfois nécessaire.
Les associations de malades fournissent accompagnement et informations au patient et à son entourage.
La maladie de Parkinson est la deuxième maladie neurodégénérative, plus fréquente dans les pays industrialisés. Elle pourrait être liée à des facteurs environnementaux (pesticides ++). Les formes génétiques représentent environ 10 % des cas.
Elle est liée à un déficit dopaminergique en lien avec une dégénérescence du locus niger ; les symptômes moteurs apparaissent lorsque 50 à 70 % de celui-ci est détruit. Avant, on parle de phase prodromale, qui peut durer de nombreuses années, avec des symptômes non moteurs (hyposmie, cauchemars, constipation...).
La symptomatologie clinique associe akinésie, rigidité, tremblement de repos (inconstant) et troubles de la marche.
Trois phases de la maladie sont décrites : la phase où les symptômes sont bien contrôlés pendant plusieurs années, les fluctuations motrices et non motrices avec l’observation de dyskinésies, et la phase de déclin moteur, cognitif et comportemental.
Les stratégies thérapeutiques sont proposées à chaque stade. D’abord, les agonistes dopaminergiques, inhibiteurs enzymatiques, dopathérapie, puis les traitements de deuxième ligne : techniques de stimulation dopaminergique continue (stimulation cérébrale profonde et pompe à apomorphine ou gel de lévodopa-carbidopa intra-intestinal), anticholinestérasiques et clozapine en y associant rééducation et orthophonie.
Dans un futur proche, les nouveaux traitements ne seront plus uniquement symptomatiques mais ils pourraient également ralentir le processus neurodégénératif : on parle du concept de neuroprotection ; plusieurs essais encourageants sont en cours (anti-alphasynucléine).
 Encadrés
Encadrés