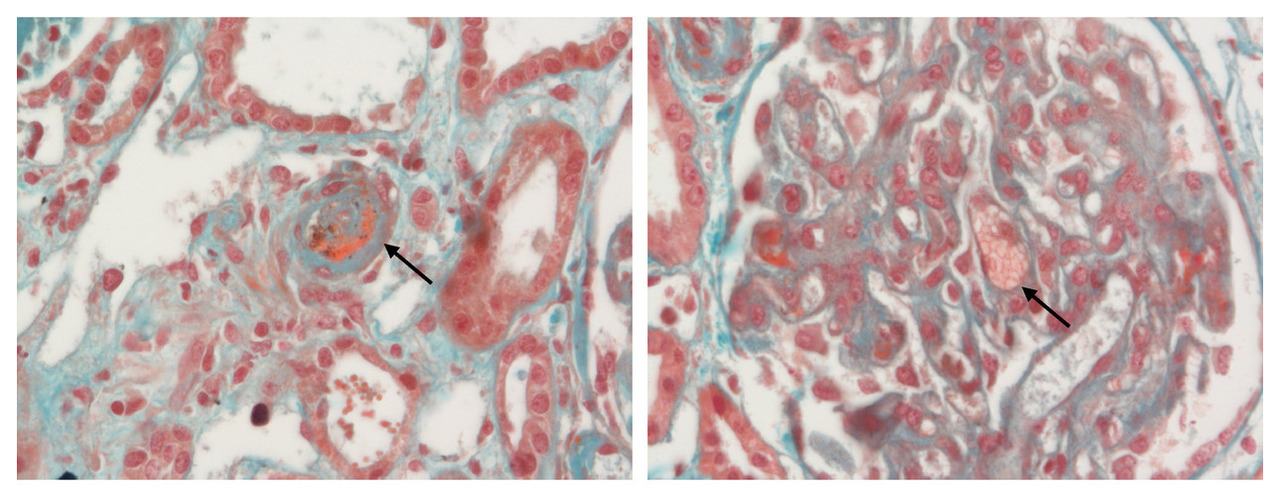
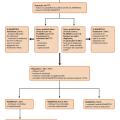
Le syndrome de microangiopathie thrombotique (MAT) est défini par l’association d’une anémie hémolytique mécanique, d’une thrombopénie de consommation et de l’atteinte d’un ou plusieurs organes en lien avec la formation de microthrombi obstruant la microcirculation (fig. 1).1 On regroupe sous ce terme un ensemble de pathologies rares, répondant à des mécanismes physiopathologiques divers. Les atteintes viscérales, de topographie et de sévérité variables, peuvent être responsables d’une importante morbi-mortalité. Le syndrome de MAT constitue donc une urgence diagnostique et thérapeutique. La classification des entités en cause a récemment évolué grâce à une meilleure compréhension des mécanismes physiopathologiques sous-jacents, avec des conséquences directes sur la prise en charge des patients.
La classification actuelle différencie les syndromes de MAT, en lien avec un mécanisme endogène dont la prise en charge repose sur un traitement ciblé, et les MAT secondaires, survenant dans un contexte particulier (infection, néoplasie, greffe, maladie auto-immune, médicament ou toxique, hypertension artérielle [HTA] sévère…), et dont la prise en charge repose surtout sur les soins de support et le traitement de l’événement ou de la pathologie associée (tableau 1).1
Purpura thrombotique thrombocytopénique
Le purpura thrombotique thrombocytopénique(PTT) est caractérisé par un déficit fonctionnel sévère (activité < 10 %) en ADAMTS13 (A Disintegrin And Metalloproteinase with ThromboSpondin type 1 repeats, 13e membre), la protéase clivant le facteur Willebrand (FW). Ce déficit a pour conséquences l’accumulation de multimères de FW de haut poids moléculaire, l’adhésion des plaquettes au FW et leur activation, donc la formation de microthrombi riches en FW au sein de la microcirculation, responsables de la triade thrombopénie de consommation, anémie hémolytique et souffrance d’organes.2 Le déficit en ADAMTS13 peut être d’origine auto-immune (95 % des cas) et lié à des anticorps anti-ADAMTS13, ou congénital (5 % des cas) en lien avec des mutations bialléliques du gène ADAMTS13.2 Il s’agit d’une pathologie rare touchant principalement l’adulte (95 % des cas), avec une incidence estimée à 1 ou 2 cas par million de personnes et par an.1,2 Dans la forme auto-immune, le PTT survient plus souvent chez la femme (sex-ratio 2 :1). L’âge médian au diagnostic est de 43 ans.2 Des facteurs intercurrents (typiquement une infection) peuvent favoriser une activation endothéliale et ainsi déclencher un épisode de la maladie.
Clinique : atteintes cardiaque et neurologique
Le tableau clinique du PTT se caractérise par une atteinte neurologique (confusion, déficit focal, convulsions, voire coma) et cardiaque (allant d’une élévation isolée de la troponine au choc cardiogénique, en passant par les troubles du rythme), souvent prédominante alors que l’atteinte rénale est absente ou modérée (créatininémie généralement inférieure à 200 µmol/L).
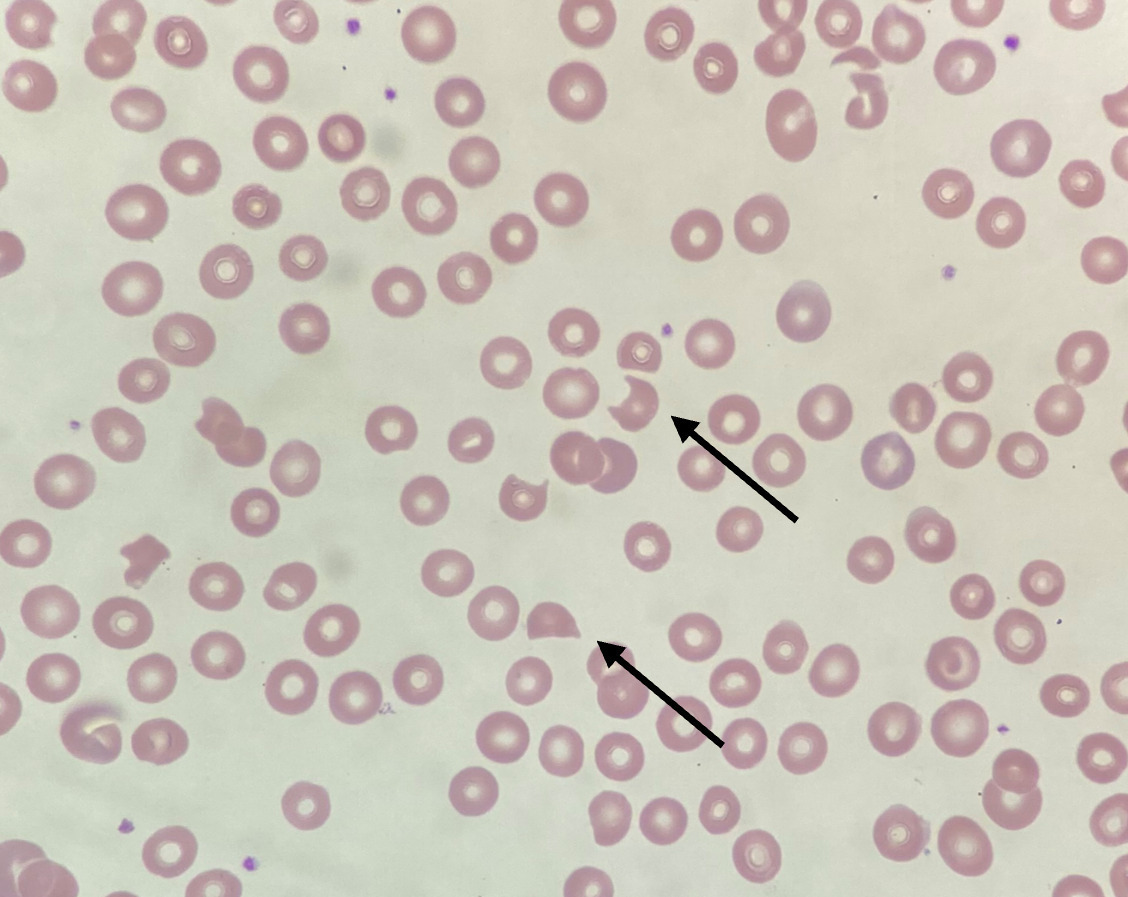
La thrombopénie est généralement profonde (inférieure à 30.109/L) ; l’anémie est régénérative ; elle s’accompagne d’une élévation des enzymes lactate déshydrogénase (LDH) [traduisant surtout les souffrances d’organes] et de la bilirubine libre, ainsi que d’un taux d’haptoglobine généralement bas ou indosable, témoignant de l’hémolyse. La présence de schizocytes au frottis sanguin signe le caractère mécanique de l’anémie hémolytique (fig. 2). Leur apparition est parfois tardive et doit justifier une recherche répétée quand ils sont absents initialement, alors que le reste du tableau clinico-biologique est évocateur. Le test direct à l’antiglobuline est classiquement négatif. Des anticorps antinucléaires, le plus souvent à faible titre, peuvent s’observer chez près de la moitié des patients et témoignent indirectement du caractère auto-immun de la maladie.
Le dosage de l’activité ADAMTS13 est indiqué en première intention pour confirmer ou infirmer le diagnostic de PTT. Il doit être réalisé avant toute transfusion ou échange de plasma (apport exogène d’ADAMTS13). Un déficit sévère en ADAMTS13 (activité inférieure à 10 %) associé à la présence d’anticorps anti-ADAMTS13 confirme le diagnostic de PTT auto-immun ; à l’inverse, un déficit sévère persistant en ADAMTS13 sans anticorps anti-ADAMTS13 détectables à distance de l’épisode inaugural doit faire réaliser des explorations génétiques dans l’hypothèse d’un PTT congénital.
Traiter en urgence
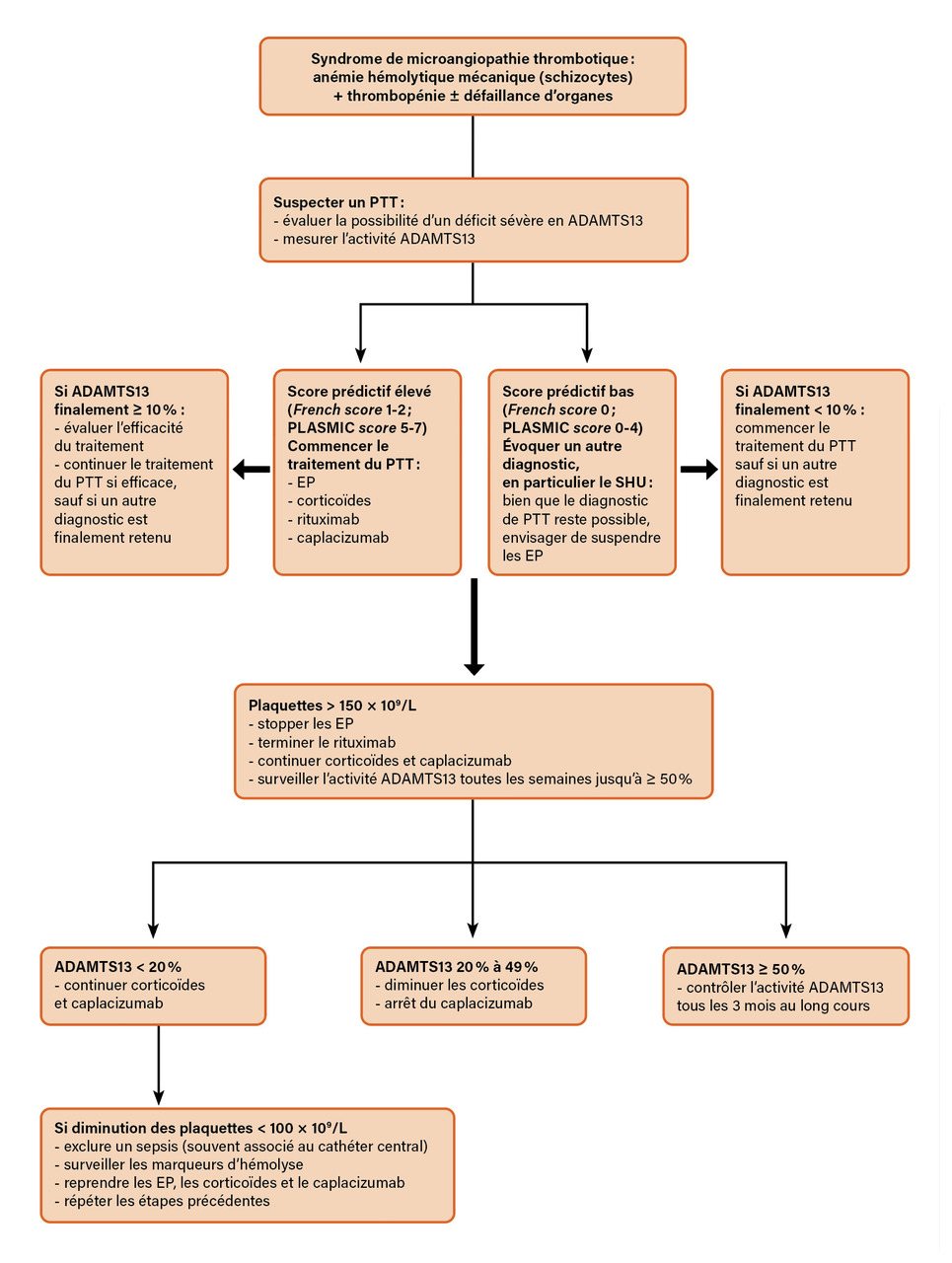
Le traitement du PTT est une urgence. Le résultat du dosage d’ADAMTS13 étant fréquemment obtenu après un délai de quelques jours, le traitement doit être initié de façon probabiliste. Des scores diagnostiques ont été proposés afin d’estimer la probabilité clinique de PTT et d’aider à la décision thérapeutique ; les plus utilisés sont le French score et le PLASMIC score (tableau 2).2
PTT auto-immun
Il peut s’associer à différents contextes favorisant sa survenue : grossesse, maladie auto-immune systémique (diagnostiquée au préalable ou plus rarement de manière concomitante), infection par le virus de l’immunodéficience humaine (VIH) à la phase initiale et plus rarement médicaments (ticlopidine, lénalidomide ou inhibiteurs de point de contrôle immunitaire).3,4
Le traitement du PTT auto-immun repose sur une triade comprenant échanges plasmatiques, immunosuppression par corticoïdes et rituximab, et inhibition de l’interaction plaquettes-FW par le caplacizumab (fig. 3).5 Les échanges plasmatiques, en remplaçant des volumes importants de plasma, permettent d’apporter une grande quantité d’ADAMTS13 sans risque de surcharge volémique voire de diminuer le titre d’anticorps anti-ADAMTS13. Ils sont poursuivis jusqu’à correction de la thrombopénie.2 Le caplacizumab est un nanocorps inhibant l’interaction FW-plaquettes. Il permet de diminuer considérablement le nombre d’échanges plasmatiques en induisant une réponse clinique rapide et durable. Il n’a pas d’activité sur la composante auto-immune ni sur l’activité ADAMTS13 et doit donc être poursuivi jusqu’à ce que l’immunomodulation permette un rétablissement de l’activité ADAMTS13 à un seuil supérieur ou égal à 20 %, suffisant pour empêcher l’apparition des manifestations du PTT.2 L’efficacité du caplacizumab est telle qu’à l’heure actuelle des stratégies thérapeu-tiques associant caplacizumab et immunosuppression sans échanges plasmatiques commencent à être envisagées, en particulier pour les formes les moins sévères de la maladie (caractérisées par l’absence de défaillance multiviscérale). L’immunomodulation associe une corticothérapie à forte dose à une déplétion lymphocytaire B par le rituximab et permet, d’une part, d’améliorer le délai de réponse par rapport aux échanges plasmatiques seuls et, d’autre part, d’éviter les rechutes durant les douze à dix-huit mois suivant l’épisode initial.2
Avec les stratégies thérapeutiques actuelles, le pronostic du PTT autoimmun est excellent, à condition que le diagnostic soit posé rapidement. Les rechutes cliniques sont concomitantes de la récupération lymphocytaire B, soit en général douze à dix-huit mois après le traitement par rituximab. La surveillance de l’activité ADAMTS13 permet d’identifier la récidive du déficit qui précède quasi systématiquement la rechute clinique. Les rechutes biochimiques (dites « ADAMTS13 ») doivent bénéficier d’un traitement préemptif par rituximab, permettant de prévenir la majorité des rechutes cliniques.2
PTT héréditaire
Le PTT héréditaire est une maladie autosomique récessive évoluant par poussées. Près de 200 mutations du gène ADAMTS13 ont été rapportées. La première poussée de la maladie a lieu en général avant l’âge de 10 ans et, dans plus de la moitié des cas, dès la naissance. Chez le nouveau-né, l’hémolyse et la thrombopénie inexpliquées motivent parfois une exsanguino-transfusion. Au début, les poussées sont totalement régressives, mais, après quelques années d’évolution sans traitement prophylactique, peuvent apparaître une insuffisance rénale chronique et d’autres défaillances viscérales chroniques, en particulier cérébrales (accidents vasculaires cérébraux, épilepsies, troubles cognitifs) et cardiaques, liées aux épisodes ischémiques répétés.
L’apport exogène d’ADAMTS13 par plasmathérapie représentait jusqu’à récemment la seule modalité thérapeutique disponible dans le PTT héréditaire. La mise à disposition dans le cadre d’un accès précoce depuis février 2024 d’une forme recombinante de la protéine ADAMTS13 constitue désormais une alternative thérapeutique avantageuse qui est amenée à remplacer la plasmathérapie historique.6 En effet, en permettant d’apporter de plus grandes quantités d’ADAMTS13, cette stratégie prévient plus efficacement les rechutes de la maladie tout en induisant moins d’effets indésirables. Elle permettra probablement de mieux protéger le patient des complications viscérales à long terme. De plus, ce traitement peut être administré à domicile.6
Syndrome hémolytique et urémique atypique
Le syndrome hémolytique et urémique (SHU) se distingue du PTT par un tropisme surtout rénal avec, à l’examen histologique, des lésions endothéliales et de nombreux microthrombi riches en fibrine.
Le terme « atypique » fait référence à l’absence habituelle de diarrhée, par opposition au SHU dit « typique » associé à une souche d’Escherichia coli sécrétrice de shigatoxine, détaillé ci-après. L’emploi de ce terme est discutable étant donné le grand recouvrement clinique entre ces deux entités et il paraît plus approprié d’utiliser les termes « SHU associé au complément (plutôt que SHU atypique) » et « SHU associé à E. coli producteur de shigatoxine » (plutôt que SHU typique).
SHU atypique associé au complément
Il résulte d’une dérégulation du complément, en lien avec un variant pathogène (ou parfois plusieurs) d’une protéine du complément : variant perte de fonction des gènes codant pour le facteur H, facteur I, CD46 ou la thrombomoduline ; gain de fonction des gènes codant pour le facteur B ou la protéine C3. Des gènes hybrides entre celui du facteur H et des protéines apparentées (CFHR1) ont également été décrits. Des SHU associés à des auto-anticorps ciblant le facteur H peuvent s’observer, majoritairement chez l’enfant. Les causes génétiques identifiées représentent 50 à 70 % des cas, chez l’adulte comme l’enfant ; les formes auto-immunes représentent 10 % des cas. Dans 30 % à 50 % des situations, aucun variant pathologique ni auto-anticorps n’est mis en évidence.7
L’activation incontrôlée de la voie alterne entraîne l’activation du C5 par la C5 convertase et, in fine, la formation du complexe d’attaque membranaire (qui se dépose sur la membrane endothéliale) et l’accumulation des fractions C3a et C5a du complément sur leurs récepteurs endothéliaux. Ces phénomènes aboutissent à des lésions endothéliales, à une activation plaquettaire et à la formation de microthrombi.7
SHU atypiques non médiés par le complément
Certains SHU atypiques sont liés à des variants sur des gènes en dehors du système du complément. Des variants perte de fonction de DGKε (diacylglycérol kinase ε) sont associés à des formes exclusivement pédiatriques survenant avant l’âge d’un an.7
Une autre forme de SHU atypique résulte d’anomalies du métabolisme intracellulaire de la vitamine B12 (déficit en cobalamine C chez l’enfant et le jeune adulte, déficit en cobalamine G chez l’enfant).7
Clinique
Le SHU atypique a une incidence annuelle de 0,25 à 2 cas par million.7 L’insuffisance rénale aiguë est au premier plan de la présentation clinique, avec nécessité d’épuration extrarénale dans la moitié des cas environ.
Elle s’accompagne volontiers d’une hypertension artérielle sévère voire maligne, susceptible d’aggraver l’hémolyse. La fraction C3 du complément est abaissée dans 20 à 40 % des cas seulement, et aide assez peu au diagnostic. La thrombopénie est généralement modérée, voire absente dans 10 à 15 % des cas. Des manifestations extrarénales sont présentes dans 20 à 40 % des cas, en particulier une atteinte neurologique, qui peut s’intégrer dans le cadre d’un syndrome d’encéphalopathie postérieure réversible (PRES) mais également une symptomatologie cardiaque (myocardite) ou digestive (diarrhées, douleurs abdominales).7
Traitement
Le traitement du SHU atypique repose sur l’éculizumab, un anticorps monoclonal humanisé ciblant la fraction C5 du complément et empêchant la formation du complexe d’attaque membranaire. L’éculizumab a une activité suspensive, son arrêt se soldant par une rechute dans 25 à 30 % des cas, a fortiori lorsqu’un variant pathogène a été identifié. Sa demi-vie est de onze jours, imposant une administration toutes les deux semaines. Un inhibiteur du C5 à longue demi-vie, le ravulizumab, autorise un plus grand espacement des doses (toutes les huit semaines). La tolérance est bonne, mais marquée par une vulnérabilité accrue aux infections à germes encapsulés, et en particulier à méningocoque, imposant une vaccination associée à une antibioprophylaxie. Il peut être administré de façon sûre pendant la grossesse.
Les échanges plasmatiques ne sont pas indiqués, sauf dans les formes associées aux anticorps anti-facteur H, où ils sont adjoints au traitement immunosuppresseur.7
SHU à shigatoxines : un exemple de SHU post-infectieux
Le SHU à shigatoxines, survient en particulier chez le jeune enfant (âge médian de deux ans). Chez l’adulte, il concerne principalement les patients atteints de multiples comorbidités, avec un pronostic défavorable dans cette population. Les sérotypes d’E. coli les plus classiquement rencontrés sont E. coli O26 et O157 :H7 (ces deux souches sont observées dans la moitié des SHU environ). La contamination s’effectue par voie alimentaire (produits laitiers, viande de bœuf, charcuterie), par contact direct ou indirect avec les animaux excréteurs et parfois par transmission interhumaine. La thrombopénie et l’insuffisance rénale surviennent à la résolution des symptômes digestifs. Une atteinte cérébrale ou cardiaque est possible. Le diagnostic repose sur la mise en évidence de shigatoxines par polymerase chain reaction (PCR) dans les selles, l’identification des souches toxinogènes sur milieu de culture et éventuellement la recherche d’anticorps dirigés contre les sérogroupes imputables.7
La prise en charge repose sur un traitement de support, avec réhydratation, et, si nécessaire, épuration extrarénale. Une antibiothérapie par antibiotique bactéricide (quinolones, bêtalactamines) serait susceptible d’aggraver le tableau clinique par lyse bactérienne libérant les shigatoxines, tandis qu’un antibiotique bactériostatique (azithromycine) pourrait posséder un effet antitoxinique bénéfique.
Les échanges plasmatiques et l’éculizumab n’ont pas d’efficacité clairement démontrée.7
SHU à pneumocoque
Les infections invasives à Streptococcus pneumoniæ peuvent être à l’origine de SHU, en particulier chez les jeunes enfants. Cette forme semble résulter de l’exposition par la neuraminidase produite par le pneumocoque de l’antigène de Thomsen-Friederichsen (antigène T), exprimé à la surface des érythrocytes, plaquettes et cellules endothéliales glomérulaires. Le test direct à l’antiglobuline peut être positif. La transfusion de culots globulaires déplasmatisés est préférée, et la transfusion de plasma est à éviter en raison de la présence possible d’anticorps préformés ciblant l’antigène T. Le traitement repose avant tout sur celui de l’infection.7
Microangiopathies thrombotiques secondaires
Les MAT secondaires sont parfois appelées à tort « SHU secondaires » par certains auteurs en raison d’une atteinte rénale fréquente et souvent au premier plan. Pourtant, leur physiopathologie et leur pronostic diffèrent grandement du SHU atypique et le rôle du complément dans la physiopathologie de ces formes n’est pas clairement démontré.
MAT associées aux cancers
Un syndrome de MAT peut survenir chez un patient atteint de cancer soit en raison de l’exposition à certains antinéoplasiques, soit en lien avec le cancer lui-même (MAT paranéoplasiques). Les cancers les plus pourvoyeurs sont les adénocarcinomes (notamment gastriques, mammaires, pulmonaires et prostatiques) avec un volume tumoral élevé, en particulier en présence de métastases osseuses avec envahissement médullaire, voire d’une leucémie carcinomateuse.8 L’activité ADAMTS13 est alors normale ou modérément abaissée. La présence d’une coagulation intravasculaire disséminée (CIVD) et/ou d’une érythromyélémie est très évocatrice du diagnostic. Une présentation respiratoire isolée est possible (MAT tumorale pulmonaire). Le pronostic est sombre, sous-tendu par l’évolution de la néoplasie elle-même. Une régression du syndrome de MAT, le plus souvent partielle, est possible en cas de bon contrôle tumoral.
Les échanges plasmatiques et l’administration d’éculizumab n’ont pas d’efficacité.8
MAT associées aux allogreffes de cellules souches hématopoïétiques
Plusieurs événements peuvent être à l’origine d’un syndrome de MAT après allogreffe de cellules souches hématopoïétiques. Les traitements (anticalcineurines), le conditionnement de la greffe, les infections, la maladie du greffon contre l’hôte sont autant de facteurs d’agression endothéliale susceptibles de participer à sa survenue.9 Les MAT post-allogreffe sont généralement de mauvais pronostic. Le traitement repose sur l’éviction des facteurs déclenchants, avec une possible efficacité du défibrotide et surtout des inhibiteurs du complément (éculizumab et, plus récemment, narsoplimab, un inhibiteur de la voie des lectines).
MAT associées aux maladies auto-immunes systémiques
Un syndrome de MAT peut compliquer plusieurs maladies auto-immunes systémiques, en particulier le lupus systémique, le syndrome des antiphospholipides (SAPL) et la sclérodermie systémique.10 La survenue d’un tableau de MAT ressemblant à un SHU, avec présentation clinique dominée par l’insuffisance rénale, témoigne en général de l’activité de la maladie auto-immune sous-jacente.11 L’éculizumab et les échanges plasmatiques n’ont pas d’efficacité démontrée, et le traitement repose essentiellement sur celui de la maladie auto-immune associée. Certaines situations particulières méritent d’être distinguées :
- une maladie auto-immune peut s’associer à un PTT auto-immun. Il faut donc débuter un traitement probabiliste du PTT si la probabilité clinique de PTT est élevée en attendant le résultat de l’activité ADAMTS13 ;
- la crise rénale sclérodermique se manifeste généralement comme un syndrome de MAT avec une insuffisance rénale et une HTA sévère chez un patient ayant une sclérodermie inaugurale ou récemment diagnostiquée, généralement avec une atteinte cutanée extensive et/ou rapidement progressive. Les anticorps anti-ARN polymérase III sont fréquemment retrouvés. Le traitement repose sur celui de la sclérodermie, associé aux inhibiteurs de l’enzyme de conversion, qui doivent être introduits le plus précocement possible et rapidement majorés jusqu’à la dose maximale tolérée ;
- le syndrome catastrophique des antiphospholipides (SCAPL) se caractérise par la survenue de thromboses microvasculaires multiples. Un syndrome de MAT est présent dans 25 % des cas. Le tableau clinique est dominé par l’atteinte rénale hypertensive, mais les atteintes cardiaque, neurologique centrale, respiratoire et cutanée sont aussi fréquemment retrouvées. La présence d’un anticoagulant circulant est quasi constante. La prise en charge repose sur l’association d’échanges plasmatiques thérapeutiques, d’une corticothérapie et d’une anticoagulation curative.11
MAT d’origine toxique ou médicamenteuse
Les syndromes de MAT d’origine toxique ou médicamenteuse (tableau 3)12 peuvent survenir par un mécanisme immuno-allergique médié par des anticorps dirigés contre un complexe entre la molécule et les plaquettes (typiquement la quinine), par une toxicité directe de la molécule sur l’endothélium (mitomycine C, gemcitabine) avec dans ce cas un mécanisme dose-dépendant ou en altérant l’homéostasie vasculaire (thérapies anti-VEGF, inhibiteurs du protéasome). L’éviction médicamenteuse est la règle. La place des inhibiteurs du complément dans cette indication est incertaine. Par ailleurs, la survenue d’un authentique PTT auto-immun a également été associée à certains médicaments (ticlopidine, lénalidomide, inhibiteurs de point de contrôle immunitaire).3,4
MAT au cours de la grossesse
Le HELLP syndrome (Hemolysis, Elevated Liver enzymes and Low Platelets) est une MAT spécifique de la grossesse définie par l’association d’une anémie hémolytique mécanique, d’une thrombopénie inférieure à 100.109/L et d’une cytolyse hépatique. Il survient dans le cadre d’une prééclampsie et s’associe donc, dans la plupart des cas, à une hypertension artérielle et à une protéinurie, après la 20e semaine d’aménorrhée.
Le traitement repose sur l’interruption de grossesse et la délivrance, avec une disparition rapide des signes de MAT (en moins de quarante-huit heures) après l’expulsion du placenta.13 La persistance ou l’aggravation des signes de MAT et d’une insuffisance rénale après le troisième jour suivant la délivrance doit faire évoquer le diagnostic de SHU atypique médié par le complément et faire débuter un traitement par éculizumab.13
Par ailleurs, un PTT peut survenir à tout moment de la grossesse. L’absence de cytolyse hépatique et une thrombopénie très profonde sont des éléments pouvant orienter vers un PTT plutôt qu’un HELLP syndrome. Dans ce cas, un traitement par échanges plasmatiques et corticothérapie doit être débuté en urgence, sans attendre les résultats de l’activité ADAMTS13. Le caplacizumab et le rituximab sont relativement contre-indiqués au cours de la grossesse et ne sont donc pas utilisés en première intention. Par ailleurs, dans 25 à 50 % des cas de PTT diagnostiqués au cours d’une grossesse, le mécanisme du déficit en ADAMTS13 est congénital. Dans ce cas, les grossesses suivantes pourront être menées sans complications grâce à une prophylaxie par ADAMTS13 recombinante. Enfin, un SCAPL peut survenir dans le contexte obstétrical, dans la majorité des cas chez une patiente ayant un antécédent déjà connu de SCAPL.14
Pronostic des MAT
Les progrès dans la compréhension de la physiopathologie des MAT permettent de les classer de manière plus rationnelle en fonction de leur mécanisme physiopathologique, ce qui a abouti à la mise en place de thérapies ciblées efficaces. Le pronostic des MAT comme le PTT et le SHU atypique, est aujourd’hui excellent, à condition qu’un diagnostic précoce soit établi. Le pronostic des MAT secondaires reste encore réservé ; une meilleure compréhension de leur physiopathologie améliorerait la prise en charge.
2. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood 2017;129(21):2836-46.
3. Elessa D, Talbot A, Lombion N, et al. Development of thrombotic thrombocytopenic purpura during lenalidomide therapy: Three new cases and review of literature. Br J Haematol 2020;188(2):338-40.
4. Jacob S, Dunn BL, Qureshi ZP, et al. Ticlopidine-, clopidogrel-, and prasugrel-associated thrombotic thrombocytopenic purpura: A 20-year review from the Southern Network on Adverse Reactions (SONAR). Semin Thromb Hemost 2012;38(8):845-53.
5. Coppo P, Bubenheim M, Azoulay E, et al. A regimen with caplacizumab, immunosuppression, and plasma exchange prevents unfavorable outcomes in immune-mediated TTP. Blood 2021;137(6):733-42.
6. Scully M, Antun A, Cataland S, et al. Recombinant ADAMTS13 in congenital thrombotic thrombocytopenic purpura. N Engl J Med 2024;390(17):1584-96.
7. Michael M, Bagga A, Sartain SE, et al. Haemolytic uraemic syndrome. Lancet Lond Eng 2022;S0140-6736(22)01202–8.
8. Oberic L, Buffet M, Schwarzinger M, et al. Cancer awareness in atypical thrombotic microangiopathies. The Oncologist 2009;14(8):769-79.
9. Schoettler ML, Carreras E, Cho B, et al. Harmonizing definitions for diagnostic criteria and prognostic assessment of transplantation-associated thrombotic microangiopathy: A report on behalf of the European Society for Blood and Marrow Transplantation, American Society for Transplantation and Cellular Therapy, Asia-Pacific Blood and Marrow Transplantation Group, and Center for International Blood and Marrow Transplant Research. Transplant Cell Ther 2023;29(3):151-63.
10. Martis N, Jamme M, Bagnis-Isnard C, et al. Systemic autoimmune disorders associated with thrombotic microangiopathy: A cross-sectional analysis from the French National TMA registry: Systemic autoimmune disease-associated TMA. Eur J Intern Med 2021;93:78-86.
11. Rodríguez-Pintó I, Espinosa G, Erkan D, et al. The effect of triple therapy on the mortality of catastrophic anti-phospholipid syndrome patients. Rheumatology 2018;57(7):1264-70.
12. Reese JA, Bougie DW, Curtis BR, et al. Drug-induced thrombotic microangiopathy: Experience of the Oklahoma Registry and the BloodCenter of Wisconsin. Am J Hematol 2015;90(5):406-10.
13. Meibody F, Jamme M, Tsatsaris V, et al. Post-partum acute kidney injury: Sorting placental and non-placental thrombotic microangiopathies using the trajectory of biomarkers. Nephrol Dial Transplant Off Publ Eur Dial Transpl Assoc - Eu. Ren Assoc 2020;35(9):1538-46.
14. Gómez-Puerta JA, Cervera R, Espinosa G, et al. Catastrophic antiphospholipid syndrome during pregnancy and puerperium: Maternal and fetal characteristics of 15 cases. Ann Rheum Dis 2007;66(6):740-6.