Le purpura est une manifestation dermatologique dont la connaissance est indispensable car il peut révéler des affections systémiques pouvant mettre en jeu le pronostic vital.
La démarche diagnostique devant un purpura est la suivante :
- poser le diagnostic de purpura devant des lésions cutanées ;
- évoquer deux urgences : une thrombopénie sévère ou un purpura fulminans d’origine infectieuse ;
- après avoir éliminé ces deux affections graves, la recherche étiologique s’oriente en fonction de la nature thrombopénique ou vasculaire du purpura ;
- cette recherche étiologique se fait parallèlement à l’évaluation de signes de gravité : risque hémorragique dans le purpura thrombopénique, importance et localisation du sepsis dans les purpuras vasculaires infectieux, atteinte viscérale sévère dans les vascularites ;
- cette démarche aboutit à un traitement spécifique.
Définitions
Aspect clinique
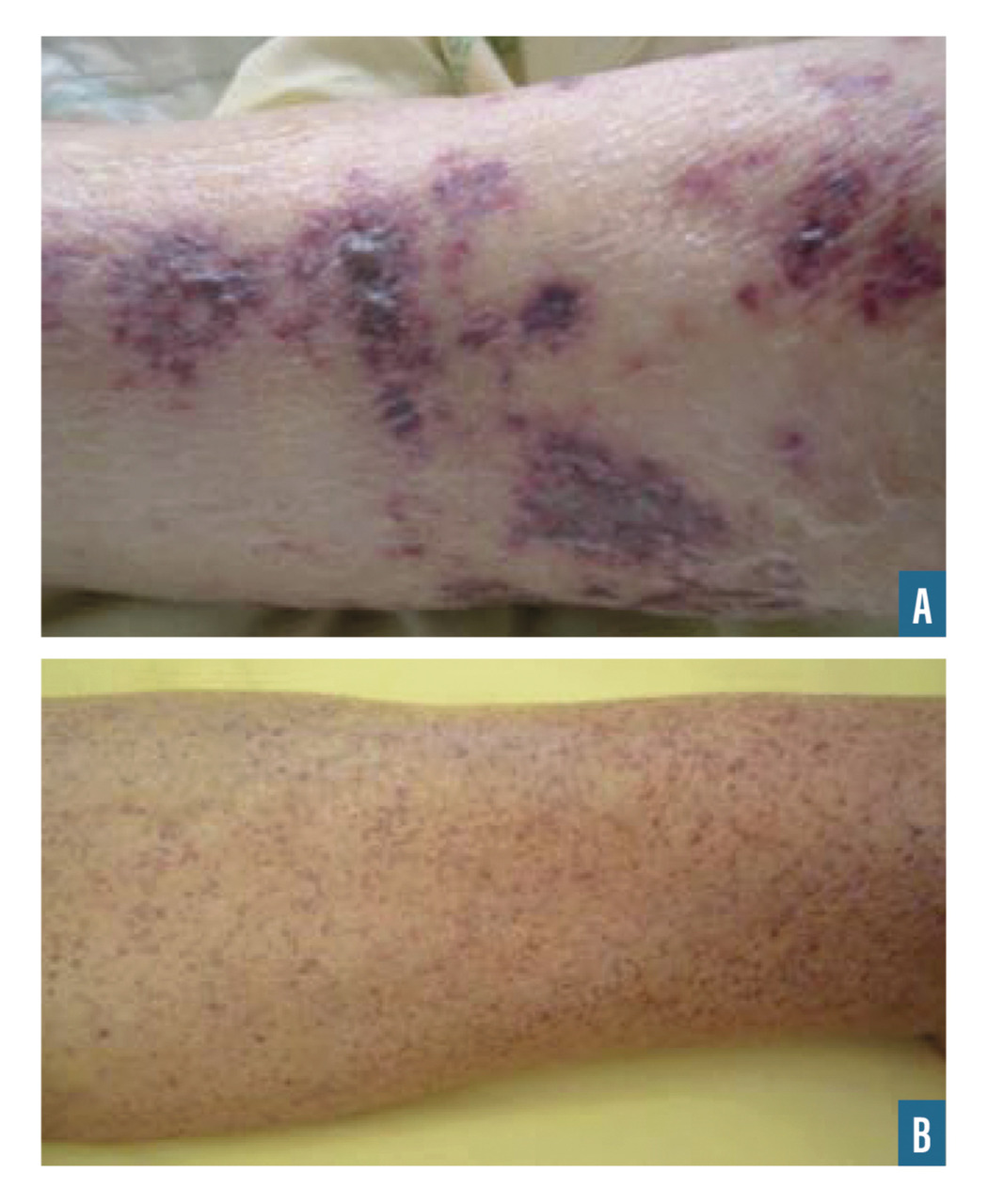
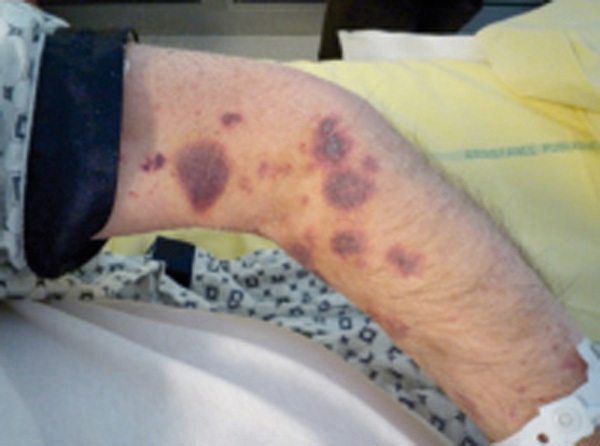
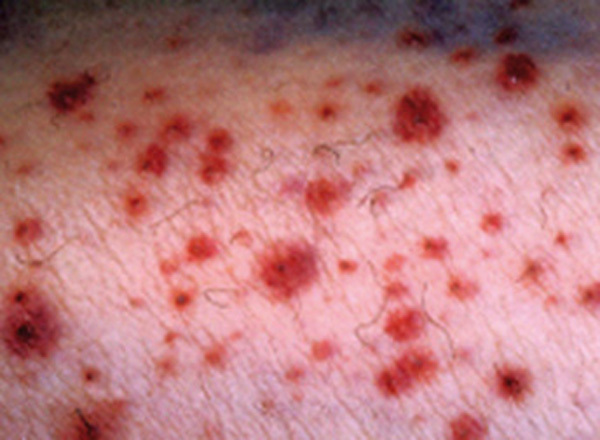
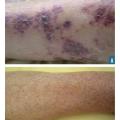
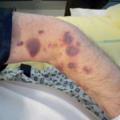
Le purpura est une lésion cutanée constituée de taches hémorragiques qui sont soit des éléments punctiformes et lenticulaires (purpura pétéchial), soit des lésions de plus grande taille (purpura ecchymotique). Les taches hémorragiques sont rouges ou violacées et sont séparées par un intervalle de peau saine. Elles ne s’effacent pas à la vitropression. Elles traduisent l’extravasation de sang hors des vaisseaux (fig. 1A et 1B).
Le purpura a deux principales origines : une pathologie de la paroi vasculaire (purpura vasculaire) ou une thrombocytopénie (purpura thrombocytopénique ou thrombopénique).
L’aspect clinique peut permettre de différencier un purpura vasculaire d’un purpura thrombocytopénique.
Le purpura vasculaire est lié à une fragilisation de la paroi vasculaire, il est le plus souvent infiltré (épaissi), associé à d’autres lésions cutanées (urticaire, bulles ou zones nécrotiques) [fig. 1A] et prédomine aux membres inférieurs. Il est aggravé par l’orthostatisme.
Le purpura thrombopénique est secondaire à un trouble de l’hémostase primaire, il est pétéchial et ecchymotique, non infiltré (fig. 1B) et peut être associé à d’autres signes hémorragiques (hématomes, ecchymoses, épistaxis, gingivorragies, bulles hémorragiques intrabuccales ou hémorragies viscérales). Bien que certaines caractéristiques cliniques puissent aider à distinguer un purpura thrombopénique d’un purpura vasculaire, seul l’hémogramme permet d’éliminer formellement un purpura thrombopénique.
Diagnostics différentiels
Il existe quatre principaux diagnostics différentiels.
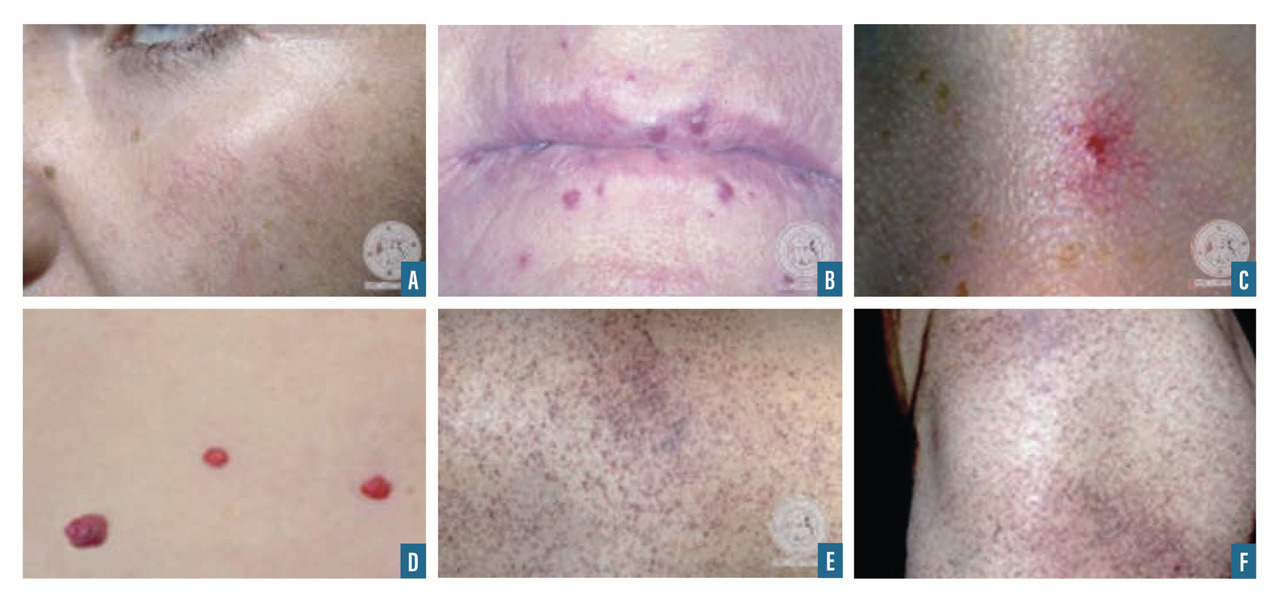
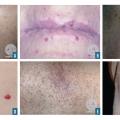
Les télangiectasies sont des dilatations des vaisseaux dermiques superficiels qui s’effacent à la vitropression (fig. 2A et B). On peut les observer dans de nombreuses affections, notamment dans la rosacée, la sclérodermie, la maladie de Rendu-Osler.
Les angiomes stellaires sont des lésions vasculaires dont l’aspect est celui d’une étoile ; leur centre est rouge avec des arborisations (fig. 2C). La pression du centre vide les arborisations. Cette lésion peut s’observer en dehors de toute pathologie mais est très évocatrice d’insuffisance hépatique lorsqu’elle est en grand nombre.
Les taches rubis sont des papules rouges ou violacées correspondant à des angiomes et n’ayant aucune signification pathologique (fig. 2D).
Les angiokératomes sont des lésions violacées infiltrées correspondant à des dilatations vasculaires (papules télangiectasiques à surface hyperkératosique) [fig. 2E et 2F]. Ils peuvent se voir dans la maladie de Fabry.
Urgences
Le diagnostic de purpura étant établi, il faut en premier lieu éliminer une urgence. Sont à rechercher systématiquement :
- une urgence infectieuse, le purpura fulminans au cours d’une infection à méningocoques, voire à pneumocoques. Il s’agit d’un purpura vasculaire ;
- une urgence hémorragique liée à une thrombopénie inférieure à 20 000 plaquettes/mm3.
Après avoir éliminé ces deux diagnostics, la démarche étiologique peut se poursuivre.
Tout purpura associé à de la fièvre doit faire immédiatement évoquer une urgence infectieuse, et en particulier le purpura fulminans.
Diagnostic étiologique
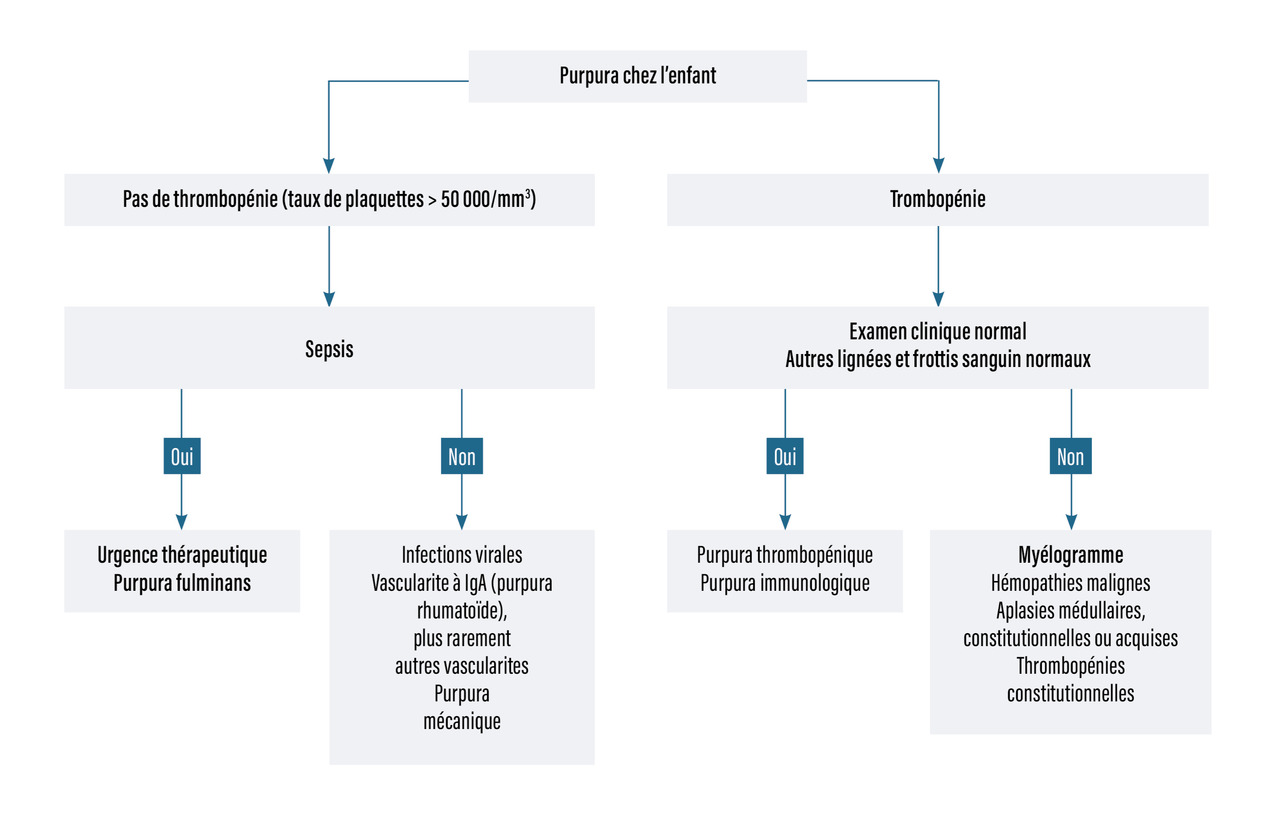
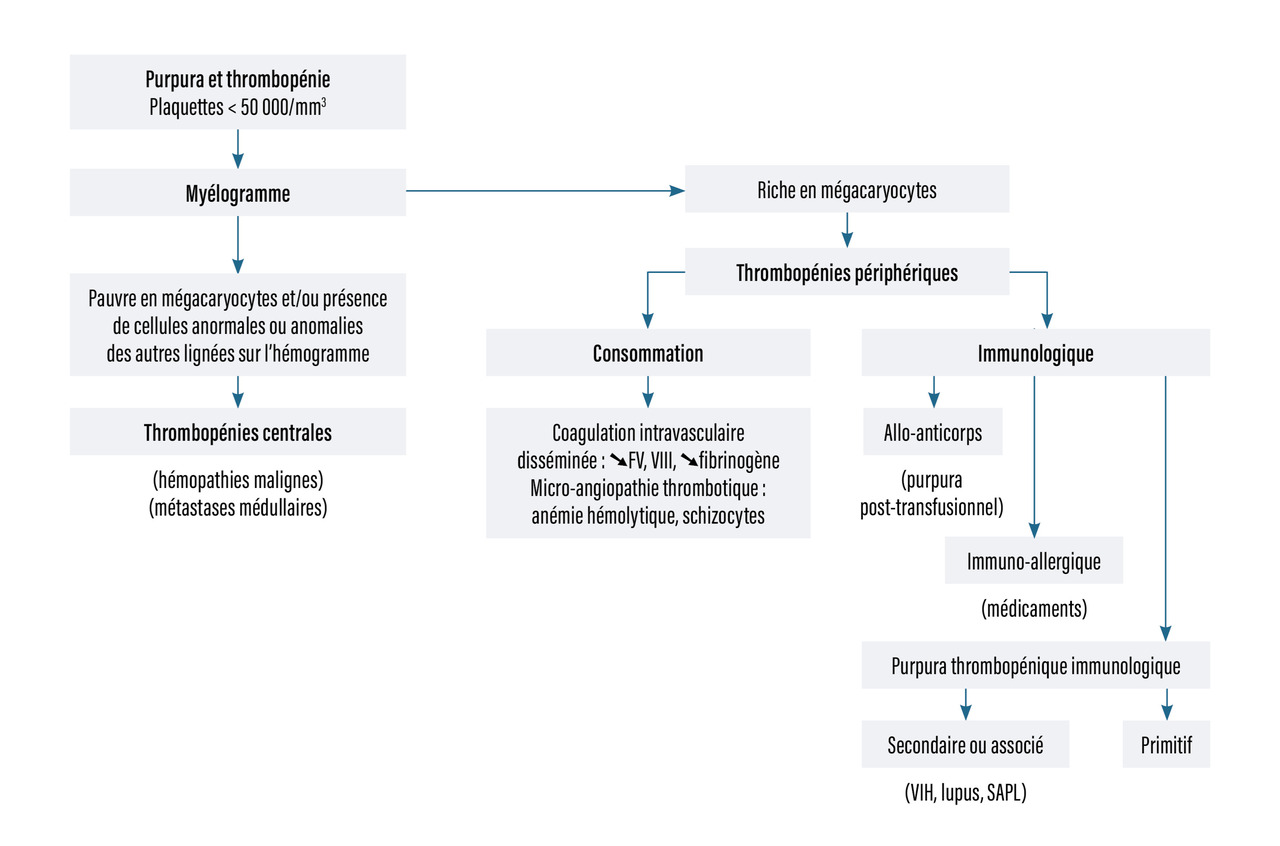
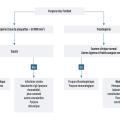
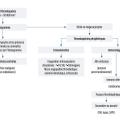
La recherche de la cause dépend de la nature du purpura, vasculaire ou thrombocytopénique (tableau 1, fig. 3).
Le contexte clinique et les manifestations associées sont des éléments d’orientation diagnostique importants (tableau 2).
Étiologie des purpuras vasculaires
Causes infectieuses
Le purpura fulminans est, chez l’adulte comme chez l’enfant, la première urgence. Le purpura fulminans est défini par l’extension rapide en taille et en nombre des éléments purpuriques, avec au moins un élément nécrotique ou ecchymotique de plus de 3 mm de diamètre, associé à un syndrome infectieux sévère. Le syndrome méningé n’est pas toujours présent. Les méningococcémies mais également les infections à pneumocoques sont en cause, elles peuvent s’accompagner de signes de coagulation intravasculaire disséminée. Une antibiothérapie par céphalosporine de troisième génération (ceftriaxone 100 mg/kg/j) doit être initiée dans les plus brefs délais.
Les endocardites peuvent se révéler par un purpura vasculaire. La présence d’un souffle cardiaque et de fièvre amène à réaliser rapidement des hémocultures et une échographie cardiaque.
Les rickettsioses (fièvre boutonneuse méditerranéenne) sont à l’origine de purpura fébrile, le plus souvent au retour de zone d’endémie.
Enfin, de nombreuses infections virales peuvent occasionner des purpuras vasculaires (parvovirus, hépatite, VIH, virus d’Epstein-Barr). Chez l’enfant, une éruption purpurique en gants et en chaussettes, souvent associée à un œdème des pieds et des mains, est évocatrice d’une infection à parvovirus B19.
Causes inflammatoires
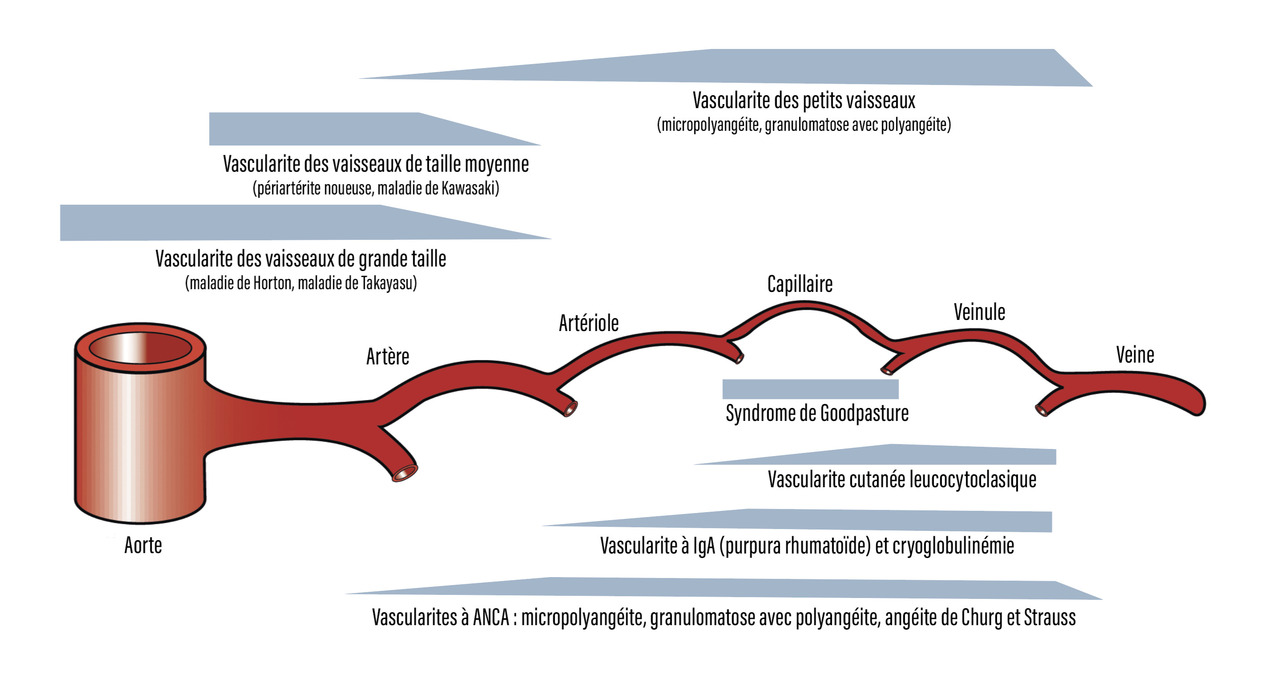
Toutes les vascularites des vaisseaux de petit et de moyen calibres peuvent être à l’origine d’un purpura vasculaire (fig. 4).
Les vascularites le plus souvent en cause sont :
- la vascularite à IgA (purpura rhumatoïde) [Henoch-Schönlein purpura] qui est une vascularite des petits vaisseaux liée à un dépôt d’immunoglobulines A ; elle est plus fréquente chez l’enfant entre 3 et 15 ans. Son incidence annuelle est estimée entre 1/6 660 et 1/4 880 chez l’enfant. La triade caractéristique associe des douleurs abdominales, des douleurs articulaires (grosses articulations des membres inférieurs) et un purpura déclive parfois très étendu. Ces signes peuvent être retrouvés de façon isolée ou apparaître successivement. Généralement, il n’y a pas de fièvre associée. Les lésions régressent spontanément en quelques jours et, dans 80 % des cas, sans récidive. Les douleurs abdominales sont communes, mais peuvent se compliquer d’hémorragie gastro-intestinale engageant le pronostic vital. Une augmentation des IgA sériques est notée dans 50 % des cas. Le traitement comporte un repos au lit, qui permet de diminuer la poussée cutanée mais n’influe pas sur la durée ou l’évolution ultérieure de la maladie. Une corticothérapie est indiquée dans les formes sévères : atteinte digestive non soulagée par le traitement symptomatique (après avoir éliminé une invagination intestinale aiguë), atteinte rénale après biopsie (recherche d’une glomérulopathie mésangiale avec dépôt d’IgA). L’atteinte rénale qui conditionne le pronostic de la maladie impose une surveillance de la protéinurie dans les mois qui suivent le diagnostic ;
- l’œdème aigu hémorragique du nourrisson qui touche l’enfant entre 6 et 24 mois. Il est caractérisé par la survenue, au décours d’un épisode infectieux viral, d’une prise de médicament ou d’une vaccination, d’un purpura papuleux parfois en cocarde, avec lésions pétéchiales pouvant devenir nécrotiques. L’éruption est associée à des œdèmes douloureux inflammatoires des extrémités dans un contexte fébrile, mais l’état général reste conservé. L’évolution est spontanément favorable en une dizaine de jours, sans séquelles. Le lien avec la vascularite à IgA (purpura rhumatoïde) de l’enfant plus grand est encore débattu.
Les vascularites à anticorps anticytoplasme des polynucléaires neutrophiles (ANCA) regroupent :
- la granulomatose avec polyangéite (anciennement granulomatose de Wegener) se caractérisant par un purpura associé à une atteinte ORL (rhinorrhée, épistaxis, sinusite ou otite) ; une atteinte pulmonaire (nodules ou hémorragie alvéolaire) ; une atteinte rénale (glomérulonéphrite extracapillaire et présence d’ANCA de type cytoplasmique à spécificité protéinase 3) ;
- l’angéite granulomateuse de Churg et Strauss qui survient fréquemment sur un terrain atopique. Elle associe un asthme, qui peut précéder le plus souvent la vascularite, des infiltrats pulmonaires, une hyperéosinophilie et l’existence d’ANCA périnucléaires à spécificité myéloperoxydase ;
- la micropolyangéite ou polyangéite microscopique qui est une vascularite des petits vaisseaux (artérioles, capillaires et veinules), se manifestant (en dehors des lésions cutanées possibles mais inconstantes) par une glomérulonéphrite pauci-immune et parfois des hémorragies alvéolaires. Les ANCA sont périnucléaires de spécificité myéloperoxydase.
La périartérite noueuse touche préférentiellement les vaisseaux de moyen et petit calibres. Elle est à l’origine d’un purpura associé à des nodules, un livedo, voire des ulcérations cutanées, des signes généraux souvent importants avec amaigrissement, fièvre, altération de l’état général, une atteinte neurologique périphérique à type de mono- ou multinévrite et des manifestations articulaires inflammatoires. L’atteinte rénale est d’origine vasculaire. Elle est responsable d’une hypertension artérielle et d’une insuffisance rénale. La recherche d’ANCA est négative.
Les cryoglobulines sont des immunoglobulines présentes dans le sérum, précipitant à froid et qui se dissolvent lors du réchauffement. Trois types sont décrits (encadré). Les cryoglobulines sont à l’origine d’un purpura vasculaire, d’un syndrome de Raynaud, de manifestations articulaires inflammatoires, d’une polynévrite et d’une glomérulonéphrite membrano-proliférative.
Les vascularites médicamenteuses (plus rares chez l’enfant) sont essentiellement responsables de manifestations cutanées à type de purpura mais sans atteinte systémique. Les médicaments le plus souvent en cause sont les sulfamides antibactériens et antidiabétiques, les bêtalactamines, les tétracyclines, les diurétiques thiazidiques, l’allopurinol, les anti-inflammatoires non stéroïdiens.
Purpuras par fragilité vasculaire
Les purpuras vasculaires ne sont pas toujours associés à une vascularite, ils peuvent aussi être liés soit à une fragilité vasculaire, soit à des dépôts, sans inflammation de la paroi vasculaire.
Le purpura sénile de Bateman est associé à une atrophie cutanée importante chez les sujets âgés (fig. 5). Un aspect superposable s’observe au cours de corticothérapie au long cours à fortes doses.
Dans le scorbut, la fragilité vasculaire peut être secondaire à une anomalie du collagène chez les patients dénutris (carence en vitamine C) [fig. 6].
Le purpura est périfolliculaire et est associé à d’autres signes hémorragiques et à une atteinte gingivale.
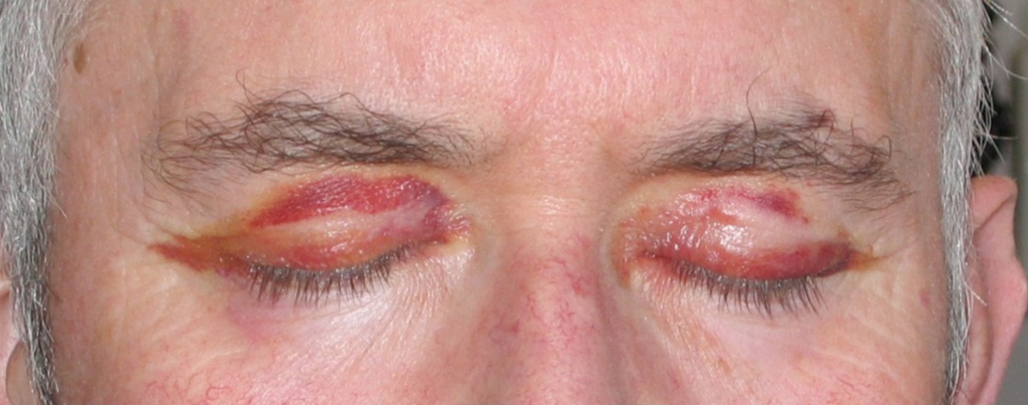
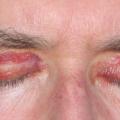
Dans l'amylose, la paroi vasculaire peut être altérée par des dépôts (fig. 7). La localisation caractéristique est périorbitaire. D’autres manifestations sont alors présentes : atteinte rénale glomérulaire, insuffisance cardiaque, neuropathie périphérique.
Le purpura mécanique peut survenir chez l’enfant, en dehors de toute pathologie vasculaire. Il s’agit d’un purpura pétéchial, limité (partie supérieure du thorax), non extensif. On retrouve à l’interrogatoire la notion d’efforts de toux, de vomissements ou de maintien d’un membre.
Étiologie des purpuras thrombocytopéniques
Les causes sont celles des thrombopénies (ou thrombocytopénies), mais toutes les thrombopénies n’entraînent pas de purpura. Le purpura ne s’observe que si les plaquettes sont inférieures à 20 000 ou 30 000/mm3. Le terme « purpura thrombopénique immunologique » a été remplacé par celui de « thrombocytopénie immune ».
Les causes des thrombopénies sont centrales ou périphériques. Le myélogramme, s’il est riche en mégacaryocytes, oriente vers une origine périphérique. Si les mégacaryocytes sont absents, il s’agit d’une thrombopénie centrale (fig. 8).
La prise en charge varie suivant l’origine de la thrombopénie.
Purpura thrombopénique immunologique
Le purpura thrombopénique immunologique (PTI) représente la première maladie auto-immune de l’enfant, caractérisée par la survenue rapide d’un purpura ou d’ecchymoses et d’une thrombopénie profonde. L’état général est conservé et l’examen clinique est toujours normal en dehors des signes hémorragiques. L’hémogramme ne montre pas d’anomalies des autres lignées et le frottis sanguin est normal. Dans ces conditions, il n’y a pas lieu de pratiquer un myélogramme. Un traitement (corticothérapie à forte dose et de courte durée ou immunoglobulines IV) est envisagé en cas de thrombopénie inférieure à 10 000/mm3 et/ou de saignement actif. La réponse thérapeutique renforce le diagnostic. Chez l’adulte, le myélogramme est recommandé au-delà de l’âge de 60 ans. L’évolution est le plus souvent chronique.
Thrombopénie par consommation
En cas de purpura avec thrombopénie peu profonde, il faut rechercher un trouble de l’hémostase associé (coagulation intravasculaire disséminée [CIVD]) ou des arguments pour une microangiopathie thrombotique (syndrome hémolytique et urémique, purpura thrombotique thrombocytopénique). Chez l’enfant, un contexte de diarrhée fébrile, une anémie hémolytique associée, une insuffisance rénale, la présence de schizocytes au frottis sont des éléments évocateurs du diagnostic de syndrome hémolytique et urémique. Chez l’adulte, il s’agit plutôt d’un purpura thrombotique thrombocytopénique, l’insuffisance rénale est plus rare et il existe un déficit en métalloprotéases ADAMTS13.
Thrombopénie centrale
La recherche d’une thrombopénie centrale et l’orientation diagnostique imposent la réalisation d’un myélogramme.
Chez l’enfant, le myélogramme doit être effectué devant des symptômes pouvant évoquer une leucémie aiguë : altération de l’état général, douleurs osseuses, anomalies des autres lignées, et avant de démarrer une corticothérapie pour une thrombopénie immunologique. Cependant, une leucémie aiguë est exceptionnellement découverte devant une thrombopénie isolée. En revanche, d’autres pathologies médullaires peuvent être à l’origine d’une thrombopénie centrale (amégacaryocytose, aplasies médullaires constitutionnelles ou acquises).
Une thrombopénie constitutionnelle peut être révélée par des signes hémorragiques si une thrombopathie est associée : syndrome MYH9 (plaquettes géantes), maladie de Willebrand de type 2B… Les antécédents familiaux de thrombopénie permettent d’orienter le diagnostic.
Chez l’adulte, l’atteinte centrale est le plus souvent responsable d’une atteinte de plusieurs lignées et donc d’une bi- ou d’une pancytopénie.
Quels examens complémentaires demander devant un purpura ?
Il faut avant tout éliminer une urgence vitale. Une numération plaquettaire inférieure à 20 000/mm3 oriente vers un diagnostic de purpura thrombocytopénique et nécessite une prise en charge adaptée.
La normalité de la numération plaquettaire ou des plaquettes diminuées modérément dans un contexte fébrile doit faire évoquer en première intention un purpura fulminans, réaliser des hémocultures et une ponction lombaire et initier une antibiothérapie par céphalosporine de troisième génération. Dans le contexte d’un purpura fébrile en rapport avec une cause infectieuse, une thrombopénie peut être constatée dans certains cas, et signe alors une origine infectieuse périphérique par consommation et non une thrombopénie d’origine immunologique.
Les urgences ayant été éliminées, les examens complémentaires doivent permettre d’aboutir à un diagnostic étiologique, qui est fonction de la nature vasculaire ou thrombopénique du purpura (tableaux 3 à 7).
Classification des cryoglobulines
Type 1 : monoclonale, plus souvent IgM et associée à une hémopathie type maladie de Waldenström, myélome ou gammapathie monoclonale de signification indéterminée.
Type 2 : mixte, composée d’immunoglobulines de classes différentes dont l’une est monoclonale de type IgM et dirigée contre une IgG polyclonale. La principale cause est l’hépatite C.
Type 3 : correspond à l’association d’immunoglobulines de classes différentes IgG et IgM polyclonales et s’observe au cours des maladies auto-immunes mais également dans certaines hémopathies.
- Tout purpura fébrile doit faire évoquer en urgence un purpura fulminans.
- Tout purpura thrombopénique est une urgence thérapeutique du fait du risque de syndrome hémorragique.
- L'aspect clinique et les signes cutanés et extra-cutanés peuvent aider à discriminer un purpura vasculaire d'un purpura thrombopénique.
- Devant tout purpura en absence de fièvre, la réalisation d'un hémogramme en urgence permet d'éliminer un purpura thrombopénique.
- Chez un enfant avec purpura et douleurs abdominales, penser à une vascularite à IgA (purpura rhumatoïde)
Bierling P, Godeau B. Purpura thrombopénique immunologique : la révolution des biothérapies? Rev Prat 2009;59(5):606-7.
Niaudet P, Pillebout E. Purpura rhumatoïde de l'adulte et de l’enfant. Rev Prat 2008;58(5):507-11.
HAS. Purpura thrombopénique immunologique. Recommandations, 2009.
 Encadrés
Encadrés