Connaître les complications d’un syndrome inflammatoire prolongé.
Connaître les principales étiologies à rechercher devant un syndrome inflammatoire.
Définition et mécanismes de la réaction inflammatoire
La réaction inflammatoire est une réaction non spécifique de défense de l’organisme contre une agression d’origine exogène (infectieuse, physique, traumatique) ou endogène (immunologique).
Dans la majorité des cas, la réaction inflammatoire est une réponse protectrice qui facilite l’élimination de l’agent pathogène et qui stimule la réparation des tissus lésés. Néanmoins, cette réponse inflammatoire peut avoir des effets délétères sur l’organisme en cas de réponse inadaptée (maladies auto-immunes) ou de réponse persistante. De nombreuses pathologies s’accompagnent d’une réaction inflammatoire, d’intensité variable : pathologies infectieuses, cancers, maladies auto-immunes, états d’hypersensibilité, maladies thromboemboliques, athérosclérose.
La réaction inflammatoire est responsable de phénomènes locaux, les quatre signes cardinaux (rougeur, douleur, tumeur, chaleur), mais peut aussi entraîner de multiples effets cliniques systémiques et biologiques. Les effets cliniques systémiques sont principalement dus à l’action des cytokines pro-inflammatoires, les interleukines 1 et 6 (IL- 1, IL- 6) et le facteur de nécrose tumorale alpha (TNF-α). Ces cytokines sont sécrétées par les cellules de la réponse immunitaire innée (polynucléaires neutrophiles, macrophages…) qui ont été stimulées par la détection d’un signal danger. La reconnaissance du signal danger se fait via des récepteurs exprimés par ces cellules, appelés pattern recognition receptors (PRR). Ils reconnaissent des signaux danger d’origine infectieuse, les pathogen-associated molecular patterns (PAMP) ou endogènes, les danger-associated molecular patterns (DAMP). Les cytokines sécrétées très rapidement par les cellules immunitaires activées ont un effet pléiotrope responsable de signes cliniques variés : asthénie, anorexie, amaigrissement, fièvre, troubles du sommeil, sarcopénie, cachexie avec fonte musculaire, œdèmes localisés ou diffus et syndrome inflammatoire. L’arrêt de la réponse inflammatoire est un processus actif impliquant les cellules immunitaires dont les macrophages différenciés en macrophages M2 et la libération de composés anti-inflammatoires comme les résolvines.
Marqueur biologique de la réaction inflammatoire : le syndrome inflammatoire
Le syndrome inflammatoire est le marqueur biologique de la réaction inflammatoire. Il est défini par l’augmentation des protéines de l’inflammation.
Protéines de l’inflammation
Les protéines dont la concentration plasmatique est modifiée d’au moins 25 % en cas de réaction inflammatoire sont appelées protéines de l’inflammation (tableau 1).
La plupart de ces protéines sont synthétisées par le foie sous l’influence de l’IL- 6 et dans une moindre mesure de l’IL- 1 et du TNF-α. À l’exception de la transferrine, de la préalbumine (transthyrétine) et de l’albumine dont les taux sériques diminuent, celui des autres protéines de l’inflammation augmente en cas de réaction inflammatoire. Dans certaines circonstances pathologiques, quelques protéines de l’inflammation sont consommées ou détruites, ce qui peut entraîner une « fausse normalité » du dosage sérique (tableau 2). Aussi, un syndrome inflammatoire est défini par l’élévation d’au moins deux protéines de l’inflammation.
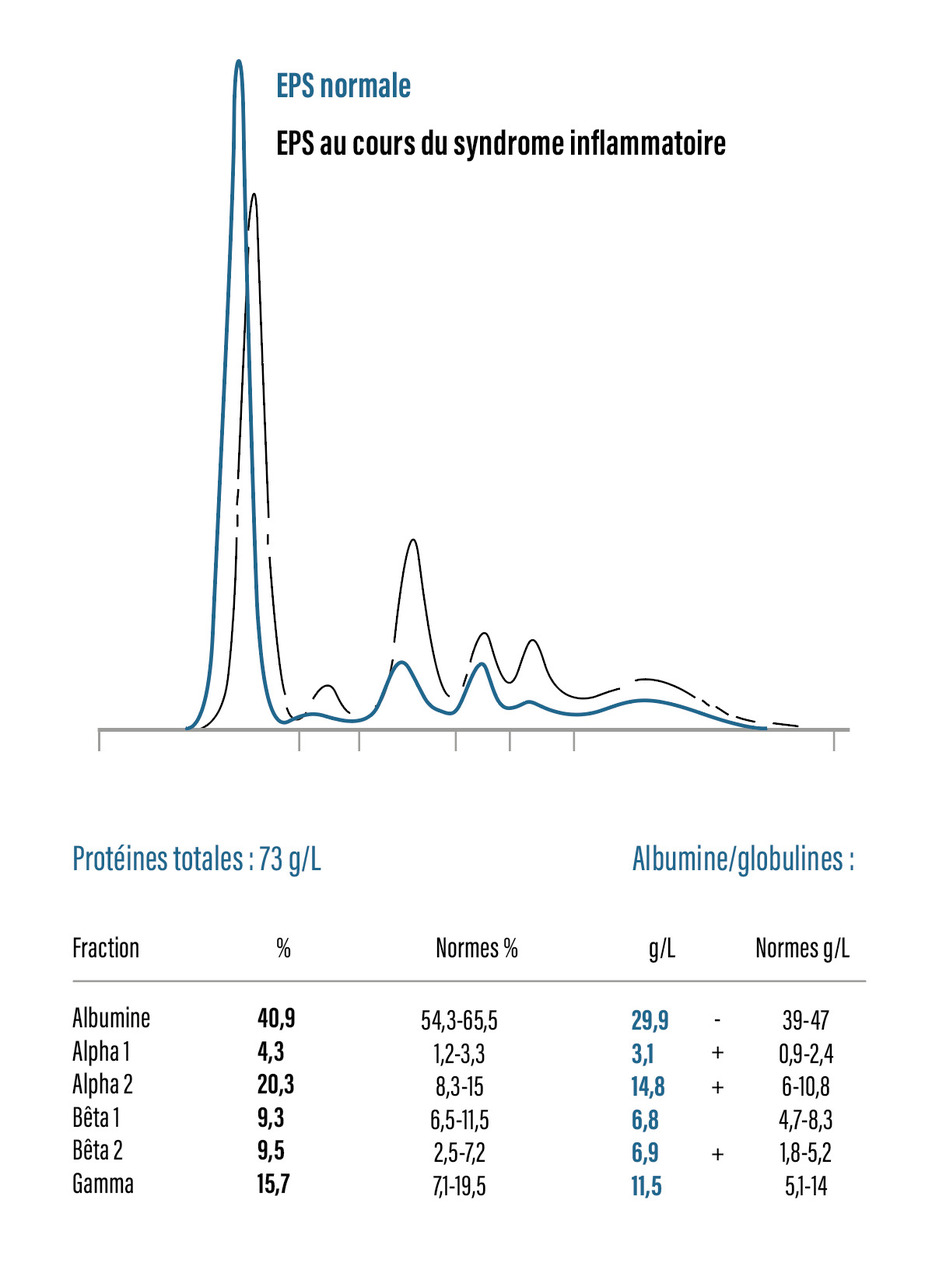
Électrophorèse des protéines sériques
L’électrophorèse des protéines sériques (EPS) permet d’individualiser cinq fractions : l’albumine, les α1 -, α2 -, β- et γ-globulines.
En cas de réaction inflammatoire, le protéinogramme montre une augmentation des pics des α1 -globulines, comprenant l’orosomucoïde et l’α1 -antichymotrypsine, des α2 -globulines, incluant l’haptoglobine et la céruloplasmine, et une diminution de l’albumine (figure).
La CRP, qui migre en β, n’influence pas le protéinogramme compte tenu de ses faibles taux sériques (mg/L), de même pour le fibrinogène, absent par définition du sérum. Les anomalies de l’électrophorèse des protéines sériques, dépendant des protéines de demi-vie longue, n’apparaissent donc que deux à quatre jours après le début de la réaction inflammatoire.
Vitesse de sédimentation
La vitesse de sédimentation (VS) était le marqueur classique du syndrome inflammatoire. C’est un test simple qui consiste à mesurer la distance parcourue par les globules rouges formant des rouleaux hématiques sédimentant dans un tube spécifique. Ce test (coté B3 [0,78 €]), moins coûteux que les autres marqueurs de l’inflammation tels la protéine C-réactive (CRP) et la procalcitonine (PCT), manque de sensibilité et de spécificité et doit être abandonné. Actuellement, pour le diagnostic comme pour le suivi précoce du syndrome inflammatoire, la VS est remplacée par la CRP, bien plus sensible et spécifique, avec une cinétique beaucoup plus rapide.
Protéine C-réactive
La CRP est une protéine de l’inflammation très sensible, appartenant à la famille des pentraxines, qui constitue un groupe de récepteurs de l’immunité innée impliquée dans la reconnaissance d’agents infectieux. La CRP est produite essentiellement par les hépatocytes sous l’action de l’IL- 6. Sa demi-vie plasmatique est courte (dix-neuf heures), une augmentation significative de sa concentration est observée six heures après le début du processus inflammatoire, et le taux de CRP peut augmenter de jusqu’à 1 000 fois sa valeur de base (< 5 mg/L). Aussi, à l’heure actuelle, il constitue le marqueur sérique le plus utile au clinicien pour confirmer et suivre un syndrome inflammatoire. Il n’existe pas de cause de non-augmentation de la CRP au cours d’une réaction inflammatoire. Lors d’une poussée de maladie lupique, son élévation est souvent faible sauf si le patient présente des épanchements, d’autres marqueurs sont alors utiles (élévation du taux des anticorps anti-ADN natif, consommation du complément). Le dosage de la CRP a donc un intérêt pour le dépistage des maladies organiques, et pour le suivi de la réponse à un traitement anti-inflammatoire ou anti-infectieux.
Procalcitonine
Au cours du sepsis, sous l’action de plusieurs cytokines (TNF-α, IL- 1β, IL- 2, IL- 6) et de dérivés bactériens, la procalcitonine est synthétisée par les cellules parenchymateuses du foie, des reins, des poumons et les adipocytes. La demi-vie de la procalcitonine est courte, de vingt à vingt-quatre heures. Sa valeur basale est inférieure à 0,1 µg/L. Ce marqueur a de bonnes spécificité et sensibilité pour discriminer une infection bactérienne d’une autre cause de syndrome inflammatoire (infections virales, maladies auto-immunes ou auto-inflammatoires…). Cependant, certaines infections bactériennes ne s’accompagnent pas toujours d’une élévation de la procalcitonine : infection localisée (appendicite non compliquée, abcès, arthrite), endocardites subaiguës, infections à germes intracellulaires, brucellose, tuberculose, borrélioses. À l’inverse, des faux positifs sont observés : cancers médullaires de la thyroïde, infections fongiques systémiques (Candida, aspergillose), infections à Plasmodium falciparum, durant les premiers jours de vie, lors de traumatismes, de chirurgie, de brûlures étendues, de coups de chaleur, au cours de l’insuffisance rénale sévère, d’une rhabdomyolyse, du choc cardiogénique et de certaines maladies auto-immunes (maladie de Still, vascularites…). L’interprétation de la procalcitonine doit toujours être confrontée aux données de l’anamnèse et de l’examen clinique.
Autres marqueurs
Protéine sérique amyloïde A
Du fait de sa demi-vie courte (24 heures) et de la grande variabilité de son taux sérique (norme < 7 mg/L), la protéine sérique amyloïde A (SAA) pourrait être un bon marqueur du syndrome inflammatoire, aussi sensible que la CRP, mais les difficultés liées à son dosage font qu’elle est peu utilisée en pratique clinique, en dehors des syndromes inflammatoires chroniques dont une des complications est l’amylose AA, liée à des dépôts de protéine amyloïde A dont la SAA est le précurseur.
Haptoglobine
Une élévation du taux d'haptoglobine de 3 à 8 fois est observée au cours du syndrome inflammatoire (normes : 0,5 - 2,5 g/L). Sa demi-vie est intermédiaire, plus longue que celle des CRP, SAA et procalcitonine mais plus courte que celle du fibrinogène. Un taux bas d’haptoglobine est observé au cours de l’hémolyse aiguë ou chronique et au cours de l’insuffisance hépatocellulaire.
Fibrinogène
Le fibrinogène est un marqueur tardif de l’inflammation du fait de sa demi-vie de l’ordre de quatre à six jours (normes : de 2 à 4 g/L). Son taux est abaissé au cours de l’insuffisance hépatocellulaire, des coagulations intravasculaires disséminées, des fibrinolyses primaires et des afibrinogénémies congénitales.
Ferritine
La ferritinémie est comprise entre 20 et 200 μg/L chez la femme et 30 et 300 μg/L chez l’homme.
De nombreux états pathologiques sont responsables d’une hyperferritinémie en dehors du syndrome inflammatoire : hémochromatose génétique, cytolyse hépatique, intoxication alcoolique chronique, insuffisance rénale chronique, maladie de Still (parfois > 10 000 µg/L) et syndromes hémophagocytaires (> 10 000 µg/L).
Modifications de l’hémogramme au cours du syndrome inflammatoire
Un syndrome inflammatoire prolongé peut entraîner des modifications de l’hémogramme :
- une anémie (de 8 à 10 g/dL), normochrome, normo- ou faiblement microcytaire, arégénérative. L’origine de cette anémie est plurifactorielle : inhibition de la production d’érythropoïétine par l’IFN-g, le TNF-α et l’IL- 1 ; augmentation de la production hépatique d’hepcidine par l’IL- 6. Cette protéine entraîne une protéolyse de la ferroportine, une protéine transmembranaire qui transporte le fer du cytoplasme vers le milieu extracellulaire. Ainsi, le fer est séquestré au sein des macrophages du système réticulo-endothélial et son absorption digestive est diminuée ;
- une thrombocytose (excédant rarement 1 million/mm3) ; l’IL- 1β et l’IL- 6 augmentent la synthèse hépatique de thrombopoïétine, principal facteur de croissance des mégacaryocytes ;
- une hyperleucocytose à polynucléaires neutrophiles (PNN) médiée par des agents exogènes bactériens et par les cytokines pro-inflammatoires. L’IL- 1 stimule la sécrétion de granulocyte-macrophage colony-stimulating factor (GM-CSF), facteur de croissance médullaire des PNN, et l’IL- 8, facteur chimiotactique des polynucléaires neutrophiles. Mais l’IL- 1 favorise aussi l’adhésion des polynucléaires aux cellules endothéliales, entraînant alors une leucopénie.
Conduite à tenir devant un syndrome inflammatoire
En pratique clinique, la découverte d’un syndrome inflammatoire est pathognomonique d’une pathologie organique. Deux situations peuvent être envisagées.
En présence d’une cause évidente de syndrome inflammatoire
C’est le cas devant une affection se présentant de façon caractéristique, que cela soit une infection (pneumonie franche lobaire aiguë, pyélonéphrite…) ou une maladie de système, par exemple l'artérite à cellules géantes. La CRP, grâce à sa cinétique rapide, permet le suivi du syndrome inflammatoire et donc de la guérison de la maladie. Devant la réascension du syndrome inflammatoire malgré un traitement étiologique bien conduit, il faut rechercher une rechute, une pathologie associée ou une complication (formation d’un abcès, thrombose veineuse profonde, arthrite microcristalline…).
En l’absence de cause évidente de syndrome inflammatoire
La démarche rejoint celle à adopter devant une fièvre prolongée (v. item n° 190). Il faut reprendre de façon minutieuse tous les antécédents médicaux, chirurgicaux, familiaux, les voyages récents, les vaccinations, les contacts avec des animaux, l’origine ethnique, la profession et les thérapeutiques. L’examen clinique doit être complet et minutieux, à la recherche de signes qui, bien que discrets, peuvent constituer des « indices diagnostiques potentiels », orientant la réalisation d’examens biologiques, d’imagerie ou une biopsie pour mener au diagnostic (tableau 3).
Causes du syndrome inflammatoire
Infections
La recherche d’une infection – en cause dans la moitié des syndromes inflammatoires – doit être systématique. Toute manœuvre invasive récente doit faire rechercher une complication iatrogène, la présence d’un corps étranger, une infection ou une thrombose de cathéter, par exemple. De même, la présence de tout matériel prothétique doit faire évoquer de principe une greffe infectieuse. Une endocardite peut être masquée par un traitement antibiotique préalable ou un traitement anti-inflammatoire. De plus, certains germes à croissance lente, parmi lesquels Abiotrophia spp. (ancien streptocoque déficient), les bactéries du groupe HACEK (Haemophilus, Actinobacillus actinomycetemcomitans, Cardiobacterium hominis, Capnocytophaga canimorsus, Eikenella corrodens, Kingella kingae) ou les levures sont difficiles à mettre en évidence. Les sérologies sont une aide au diagnostic pour les maladies à germes intracellulaires (légionellose, rickettsiose, brucellose, yersiniose, infections à Chlamydia…), la syphilis, les affections virales (CMV, EBV, VIH, hépatites B et C) et les parasitoses (anguillulose...). Des foyers infectieux profonds (pleuropulmonaires, hépatiques, sinusiens, dentaires, diverticulaires, gynécologiques, osseux, urinaires et prostatiques) peuvent être parfois difficiles à identifier quand il n’existe ni signe clinique évocateur ni orientation biologique. Leur recherche clinique doit être complétée par des examens d’imagerie. Une tuberculose doit être systématiquement évoquée.
Cancers
Les cancers à évoquer en l’absence de point d’appel évident sont principalement les hémopathies, et plus particulièrement les lymphomes. La recherche d’un cancer solide est orientée par les facteurs de risque personnels et familiaux, qui guident le choix des examens endoscopiques et d’imagerie. Néanmoins, en l’absence d’altération de l’état général, de fébricule ou de signes cliniques d’appel, il est très rare qu’un syndrome inflammatoire isolé permette la découverte d’une tumeur solide. En l’absence d’éléments d’orientation, la réalisation d’une tomographie par émission de positons (TEP) couplée à un scanner peut être utile, permettant de détecter des adénopathies actives ou une tumeur. Cependant, la TEP n’est pas très sensible pour détecter les tumeurs digestives, et l’endoscopie digestive reste l’examen de référence. Les lymphomes hodgkiniens ou non hodgkiniens peuvent être parfois de diagnostic difficile, et le recours à une biopsie médullaire, un immunophénotypage lymphocytaire sanguin avec un NGS myéloïde peut être utile même en l’absence de syndrome tumoral. Des myélodysplasies ou des syndromes plus rares type VEXAS peuvent se révéler par un syndrome inflammatoire parfois isolé, ou parfois accompagné de manifestations cliniques à type de vascularite ou de polyarthrite.
Plusieurs types de tumeurs rares peuvent sécréter de l’IL- 6 (cancer du rein, maladie de Castleman, carcinome thymique…), entraînant ainsi un syndrome inflammatoire.
Maladies systémiques
Les maladies systémiques s’accompagnent généralement d’un syndrome inflammatoire qui est cependant rarement isolé. L’interrogatoire et l’examen clinique permettent de collecter des signes qui orientent vers une pathologie précise et la réalisation d’examens secondaires discriminants. Les causes possibles regroupent les vascularites, les rhumatismes inflammatoires, les connectivites et les granulomatoses. Le diagnostic est rarement évoqué devant un syndrome inflammatoire isolé, bien qu’au cours des formes débutantes un diagnostic précis soit parfois difficile.
Après 55 ans, un syndrome inflammatoire isolé doit conduire à la réalisation d’une biopsie d’artère temporale associée à une TEP même si l’interrogatoire et l’examen clinique ne retrouvent pas de signe caractéristique d’artérite à cellules géantes (maladie de Horton).
L’élévation préférentielle de certaines protéines au cours du syndrome inflammatoire permet parfois une orientation diagnostique. En effet, les taux de ferritine les plus élevés (quelquefois à plus de 10 000 ng/mL), sont observés lors de la maladie de Still et du syndrome d’activation macrophagique (syndrome hémophagocytaire).
Pathologies vasculaires
La présentation clinique des thromboses veineuses est polymorphe et parfois asymptomatique. Le diagnostic doit être évoqué devant un syndrome inflammatoire inexpliqué, dont l’intensité est généralement proportionnelle à l’étendue de la thrombose.
L’infarctus du myocarde et les anévrismes de l’aorte s’accompagnent d’un syndrome inflammatoire mais souvent d’intensité modérée.
Autres
Les syndromes auto-inflammatoires constituent un groupe de pathologies très rares liées à diverses mutations génétiques et caractérisées par la survenue de fièvres récurrentes associées à un syndrome inflammatoire, cela en l’absence d’agents pathogènes. Ces anomalies sont liées à une dysrégulation de l’immunité innée avec production accrue d’IL- 1β et de TNF-α responsables du processus inflammatoire. Chez l’adulte, les présentations cliniques associent manifestations articulaires et, selon la pathologie considérée, inflammation des séreuses et pseudo-érysipèle au cours de la maladie périodique, ou fièvre méditerranéenne familiale (mutation du gène MEFV codant pour la pyrine), adénopathies cervicales et hépatosplénomégalie pour le syndrome hyper-IgD (mutation de la mévalonate kinase), œdème périorbitaire et pseudocellulites au cours du TNF receptor associated periodic syndrome (TRAP), urticaire et surdité du syndrome de Muckle-Wells.
L’histoire familiale, l’origine ethnique et les manifestations cliniques permettent en général d’orienter les recherches génétiques qui permettent d’aboutir au diagnostic.
La fibrose rétropéritonéale (fibrose péri-aortique et péri-iliaque), affection idiopathique ou secondaire à des médicaments, tumeurs, infections, traumatismes ou dans le cadre d’une maladie associée aux IgG4, est responsable d’un syndrome inflammatoire. La fibrose peut entraîner une compression extrinsèque des uretères réalisant un tableau de pseudocolique néphrétique ou une insuffisance rénale obstructive lorsque l’atteinte est bilatérale.
Une cause médicamenteuse doit être évoquée devant un syndrome inflammatoire inexpliqué. Généralement, il existe des signes d’orientation, comme une fièvre, une éruption cutanée, une hyperéosinophilie.
Complications
Le risque de thrombose veineuse profonde est multiplié par quatre lorsqu’il existe une élévation du fibrinogène au-dessus de 5 g/L. Une cachexie peut compliquer les syndromes inflammatoires prolongés. Elle se définit par une altération profonde de l’état général associée à une perte musculaire importante. Sous la dépendance des cytokines pro-inflammatoires, notamment le TNF-α, initialement nommé cachectine, des altérations métaboliques des protides, lipides et glucides se produisent. L’hypercatabolisme apparaît comme le mécanisme principal de la perte de poids.
Peuvent s’y associer des phénomènes d’anorexie, des altérations gastro-intestinales (altération de la motricité digestive, diminution de la perfusion sanguine) qui favorisent la malnutrition. Le syndrome inflammatoire chronique peut aussi entraîner une ostéoporose et des complications cardiovasculaires en favorisant l’athérosclérose.
L’amylose de type AA est une complication rare des syndromes inflammatoires chroniques (syndromes auto-inflammatoires, rhumatismes et entéropathies inflammatoires, suppuration chronique, cancers) liée aux dépôts systémiques de protéine sérique amyloïde. La survie à long terme dépend de la pathologie sous-jacente et de la capacité à normaliser les taux de SAA.
L’insuffisance cardiaque et surtout l’insuffisance rénale constituent les principales causes de mortalité. Le traitement repose donc surtout sur celui de la cause.
Traitements
Le traitement reste évidemment celui de la cause du syndrome inflammatoire. Rarement, se pose cependant le problème de l’indication d’un traitement d’épreuve en cas de profonde altération de l’état général lorsqu'aucune cause n’est retrouvée après réalisation d’examens complémentaires exhaustifs. Cette situation doit rester exceptionnelle.
La première crainte étant une infection non diagnostiquée, une antibiothérapie probabiliste, de large spectre, couvrant notamment les germes intracellulaires doit être discutée. Sa durée est adaptée à l’évolution clinique et biologique.
Enfin, devant l’hypothèse d’une maladie inflammatoire non précisée, la mise en route d’une corticothérapie peut être proposée. Elle n'est administrée qu’après avoir éliminé une pathologie infectieuse. La posologie proposée n’est alors pas codifiée, mais doit être suffisante pour permettre le contrôle de la pathologie inflammatoire. Avec une meilleure compréhension et un meilleur diagnostic des syndromes auto-inflammatoires grâce aux progrès de la génétique, cette situation doit être de moins en moins fréquente.
Conclusion
L’élévation des protéines de l’inflammation définit le syndrome inflammatoire. La recherche de la cause du syndrome inflammatoire doit être adaptée en fonction du tableau clinique et biologique. Les causes principales sont les infections, les cancers et les maladies systémiques. Malgré l’utilisation récente de nouveaux marqueurs biologiques, aucun n’est pathognomonique d’une cause donnée. Le traitement du syndrome inflammatoire repose sur celui de sa cause, l’instauration d’un traitement empirique doit rester exceptionnelle et n’est envisagée qu’en cas de profonde altération de l’état général après un bilan exhaustif négatif. Le suivi est important lorsque le syndrome inflammatoire isolé reste inexpliqué : il peut régresser spontanément, traduisant généralement un processus infectieux transitoire, tandis que l’apparition de signes cliniques permet de poser un diagnostic de maladie inflammatoire ou de cancer notamment.
Le syndrome inflammatoire est un marqueur biologique défini par l’augmentation de protéines de l’inflammation due à l’action de l’interleukine (IL), l’IL- 6 et du TNF-α sur les hépatocytes.
Parmi toutes les protéines de l’inflammation, la protéine C-réactive est le meilleur marqueur du syndrome inflammatoire en raison de sa courte demi-vie et de l’augmentation importante de son taux (jusqu'à mille fois sa valeur de base).
La découverte d’un syndrome inflammatoire affirme le caractère organique de la pathologie causale sans pour autant être spécifique d’une cause particulière. Les principales causes sont les maladies infectieuses, les maladies autoimmunes ou auto-inflammatoires et les cancers.
Dans la plupart des cas, un diagnostic étiologique est rapidement posé, qu’il s’agisse d’une infection ou d’une maladie inflammatoire en poussée, et l'évolution du syndrome inflammatoire est utilisée pour suivre l’efficacité thérapeutique.
La prise en charge diagnostique des patients présentant un syndrome inflammatoire sans cause évidente doit faire discuter méthodiquement le type et l’ordre des investigations complémentaires. Même si aucune cause n’est mise en évidence, un traitement symptomatique du syndrome inflammatoire doit être discuté pour éviter les complications du syndrome inflammatoire chronique, notamment l’amylose AA et la cachexie.
 Encadrés
Encadrés