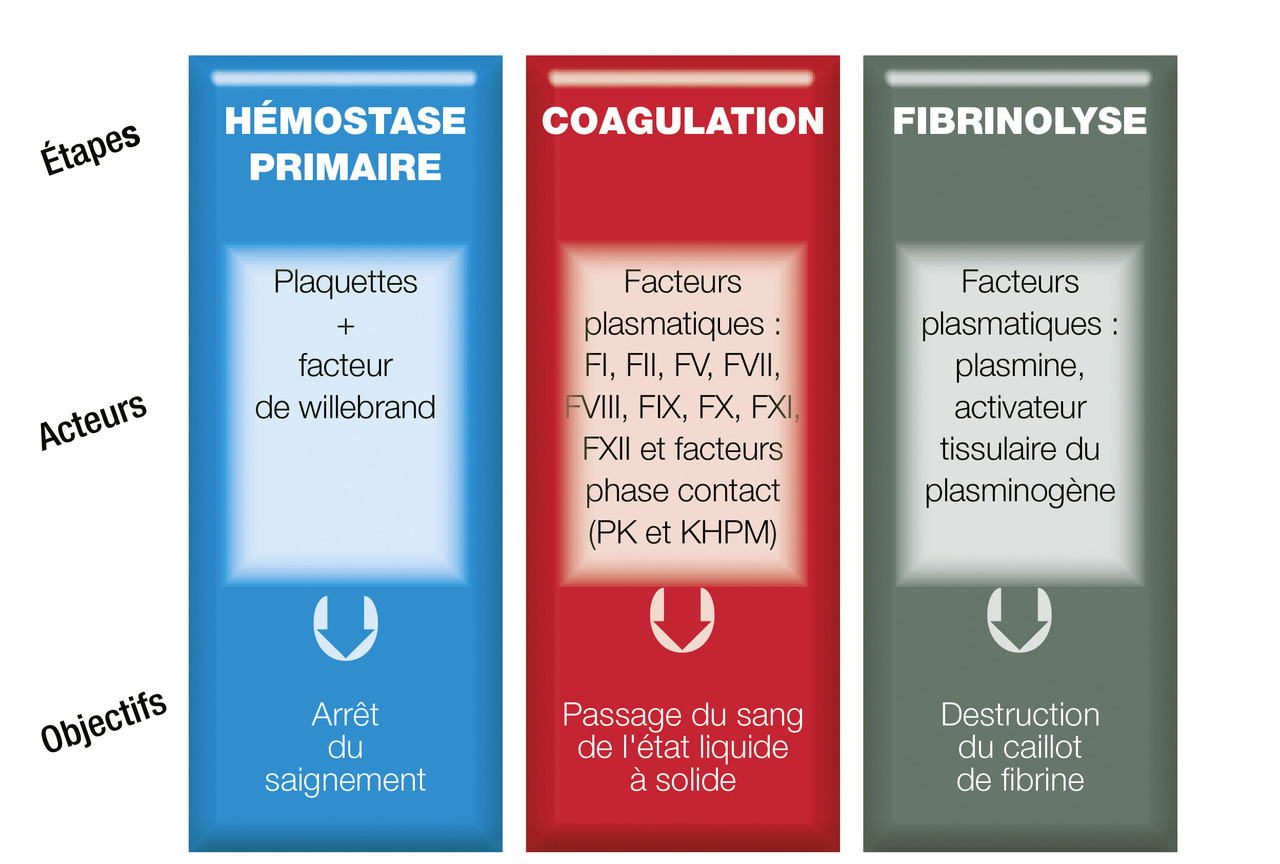
Rappels sur l’hémostase physiologique (fig. 1)
L’hémostase est le processus physiologique qui permet de limiter les pertes sanguines après une effraction vasculaire. Elle permet la constitution d’un caillot amarré au niveau de la brèche vasculaire. Ce caillot est constitué de plaquettes agrégées retenues par un filet de fibrine.
Lors d’une plaie vasculaire, l’exposition du sous-endothélium va favoriser l’adhésion des plaquettes à la paroi du vaisseau par l’intermédiaire du facteur de von Willebrand (FvW). Les plaquettes vont être activées et vont permettre la fixation du fibrinogène qui relie les plaquettes les unes aux autres. Le clou plaquettaire instable est ainsi formé.
Le sous-endothélium va libérer du facteur tissulaire qui va directement activer le facteur VII. Le facteur VII activé va enclencher la réaction de coagulation qui est une cascade de réactions enzymatiques dont le but est la transformation du fibrinogène (soluble) en fibrine (insoluble). Chaque facteur de coagulation est une enzyme ou une co-enzyme (fibrinogène-facteur I, thrombine-FII, FV, FVII, FVIII, FIX, FX, FXI, FXII, FXIII). Une fois la fibrine stabilisée, le caillot ainsi formé est stable.
La fibrinolyse est l’étape ultime permettant la dissolution du caillot et la restitution de la fluidité sanguine.
S’orienter par l’interrogatoire
La démarche diagnostique est la même quel que soit l’âge.
Il faut avant tout définir si le tableau hémorragique s’inscrit dans une pathologie connue ou associée (insuffisance hépatique ou rénale chronique, pathologie oncologique ou hémopathie, sepsis, choc hémorragique, situation obstétricale [accouchement], malformation vasculaire étendue). Nous ne traiterons pas ici de ces causes spécifiques.
En dehors d’un contexte médical spécifique, il faut alors définir le caractère primaire ou secondaire des manifestations, leurs caractéristiques et évaluer leur retentissement.
La date de début des signes hémorragiques permet de distinguer les pathologies constitutionnelles ou acquises et/ou leur sévérité : début en période néonatale, dans l’enfance, à l’âge adulte ; spontanément ou post-chirurgical ; prise en charge de ces épisodes (consultation médicale, transfusion, reprise chirurgicale).
Évaluer le caractère familial des symptômes : la présence d’antécédents hémorragiques chez les ascendants et/ou descendants/collatéraux directs sont des arguments pour une pathologie constitutionnelle ; la consanguinité favorise l’émergence de déficits rares en facteur de la coagulation.
Caractériser les manifestations cliniques aide à orienter le bilan biologique. Il est important de préciser :
- le type de manifestation : purpura cutané, ecchymoses, hématomes (sous-cutanés, profonds), hémarthroses, épistaxis, gingivorragies, ménorragies, saignements digestifs ;
- leur localisation principale : cutanéo-muqueux, musculaire, articulaire, gynécologique, mixte ;
- le caractère spontané ou provoqué, précoce ou retardé des saignements ;
- la durée des saignements.
Évaluer le retentissement de ces manifestations :
- taille des lésions cutanées, durée des épistaxis ;
- anémie associée, carence martiale connue ;
- mesures associées : traitement symptomatique, consultation, geste local, complication postopératoire.
Relever les prises médicamenteuses : faire une liste complète des traitements utilisés. Il faut penser à interroger les patients spécifiquement sur l’utilisation de compléments alimentaires et de phytothérapie (action antiagrégante). La liste de médicaments pouvant avoir des interactions secondaires avec l’hémostase est large, en dehors des plus évidents comme les anticoagulants, les antiagrégants ou les anti-inflammatoires non stéroïdiens : inhibiteurs de la recapture de la sérotonine, antidépresseurs tricycliques.
S’orienter par les signes cliniques
L’examen clinique est essentiel et oriente vers une pathologie acquise, conséquence d’une maladie associée, ou vers une pathologie constitutionnelle. Il doit également s’attacher à évaluer le retentissement clinique des manifestations hémorragiques (pâleur, tachycardie, hypovolémie…).
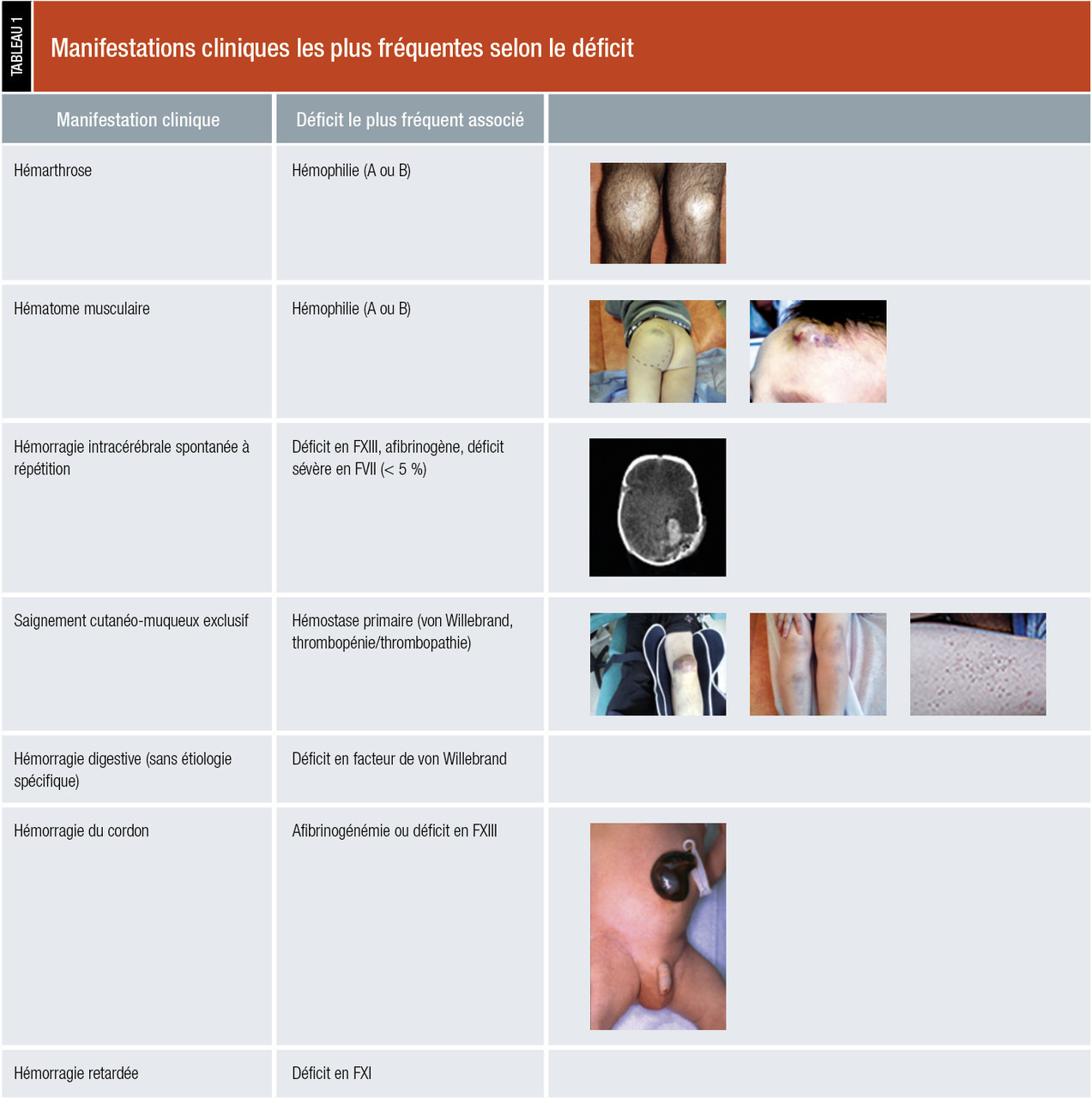
Manifestations hémorragiques (tableau 1)
L’examen doit s’attarder à rechercher toutes les manifestations hémorragiques ou leurs séquelles :
- cutanées : purpura pétéchial (diffus, localisé), localisation (zone de pression, membres inférieurs…), ecchymoses (nombre, taille < ou > 1 cm, localisation : zone exposée ou pas ; spontanée ou post-traumatique) ;
- muqueuses : nez (épistaxis), bouche (gingivorragie), pharynx, gynécologique (ménométrorragies) ;
- profondes (hématome musculaire) : douleur, taille, signes compressifs (nerf, vaisseaux, syndrome des loges) ;
- articulaires (hémarthrose) : douleur, impotence fonctionnelle, amplitudes articulaires, déformations.
Il existe des spécificités pédiatriques afin de rechercher à l’interrogatoire ou dans le carnet de santé des manifestations précoces orientant le diagnostic : les céphalhématomes (période néonatale ou enfance, spontané ou avec traumatisme minime), hématome disséquant du scalp (période néonatal sans traumatisme évident), hématome sous-périosté, hémorragie du cordon et hémorragies intracérébrales spontanées et/ou récidivantes (typiques des afibrinogénémies ou déficits sévères en FXIII).
Il faut décrire le contexte de ces manifestations :
- saignements spontanés ;
- saignements sur ponction : veineuse, vaccins ;
- saignement post-chirurgical : penser à interroger sur tous les gestes même les plus anodins comme une extraction dentaire ou une biopsie cutanée.
Signes cliniques associés orientant vers une pathologie constitutionnelle ou acquise
Il faut rechercher des signes en faveur d’une pathologie sous-jacente : insuffisance hépatique ou rénale, infection, maladie dite « de système » ou auto-immune (lupus), hémopathie maligne, cancer.
Signes de gravité évaluant le caractère de sévérité du saignement
Ce sont :
- les signes cliniques évoquant une anémie, une carence martiale, une hypovolémie ;
- les signes compressifs : vasculaire, neurologique, voies aériennes supérieures ;
- les troubles de conscience.
S’orienter par le bilan biologique
Numération formule plaquettaire
L’hémogramme permet d’éliminer les étiologies hématologiques associées à une thrombopénie et d’évaluer les signes de gravité liés à l’anémie. Nous ne traiterons pas ici les causes des thrombopénies (
En l’absence de thrombopénie, il faut orienter le bilan en fonction de l’interrogatoire et des signes cliniques vers une anomalie de l’hémostase primaire, de la coagulation ou de la fibrinolyse.
Bilan d’hémostase
Hémostase primaire
Temps de saignement (TS) : il n’est plus recommandé. Il s’agit d’un examen opérateur – et technique – dépendant, avec une reproductibilité et une sensibilité médiocres ayant abouti au déremboursement (hors nomenclature des examens biologiques).
Temps d’occlusion plaquettaire in vitro (PFA 100) : cet examen évalue la capacité fonctionnelle globale des plaquettes en sang total citraté sans préparation, en simulant une brèche de la paroi artériolaire, et réalise une hémostase artificielle. Il n’est pas recommandé en première intention. Son intérêt reste limité au dépistage de thrombopathies et déficits en FvW sévères, notamment chez l’enfant dont les prélèvements sont limités. Sa sensibilité est médiocre pour le dépistage des formes modérées de déficit en facteur de von Willebrand ou de thrombopathies (examen non remboursé car hors nomenclature des examens biologiques)
Dosages spécifiques du facteur facteur de von Willebrand : il s’agit de dosages spécifiques de l’activité ou de la molécule de facteur de von Willebrand. Le dépistage est réalisé par le dosage de l’activité du FvW appelé activité du cofacteur de la ristocétine (RCo-vWF).
Si l’activité RCo-vWF est diminuée, il est nécessaire de compléter les dosages avec la mesure antigénique du FvW et le dosage du facteur VIII (FVIII) afin de caractériser le déficit en déficit quantitatif (type 1 ou 3 dont le rapport RCo-vWF/Ag vWF > 0,7) ou en déficit qualitatif (type 2 dont le rapport RCo-vWF/AG vWF < 0,7).
Le FvW se lie au FVIII dans la circulation afin de le protéger de la dégradation. De ce fait, un allongement du TCA peut être associé au déficit en FvW en raison d’une diminution proportionnelle du FVIII.
Les tests d’agrégation plaquettaire : il s’agit de tests sur sang total réalisés en centre spécialisé. Ils doivent être discutés devant un syndrome hémorragique clinique cutanéo-muqueux inexpliqué et un bilan de coagulation standard normal.
Coagulation
Les dosages du TP, du TCA et du fibrinogène sont la base. Une anomalie d’un ou plusieurs de ces tests conduit à la réalisation de dosages spécifiques à visée diagnostique. Un dosage diminué en facteur de la coagulation nécessite toujours un contrôle avant d’affirmer un déficit.
Les tests de dépistage sont :
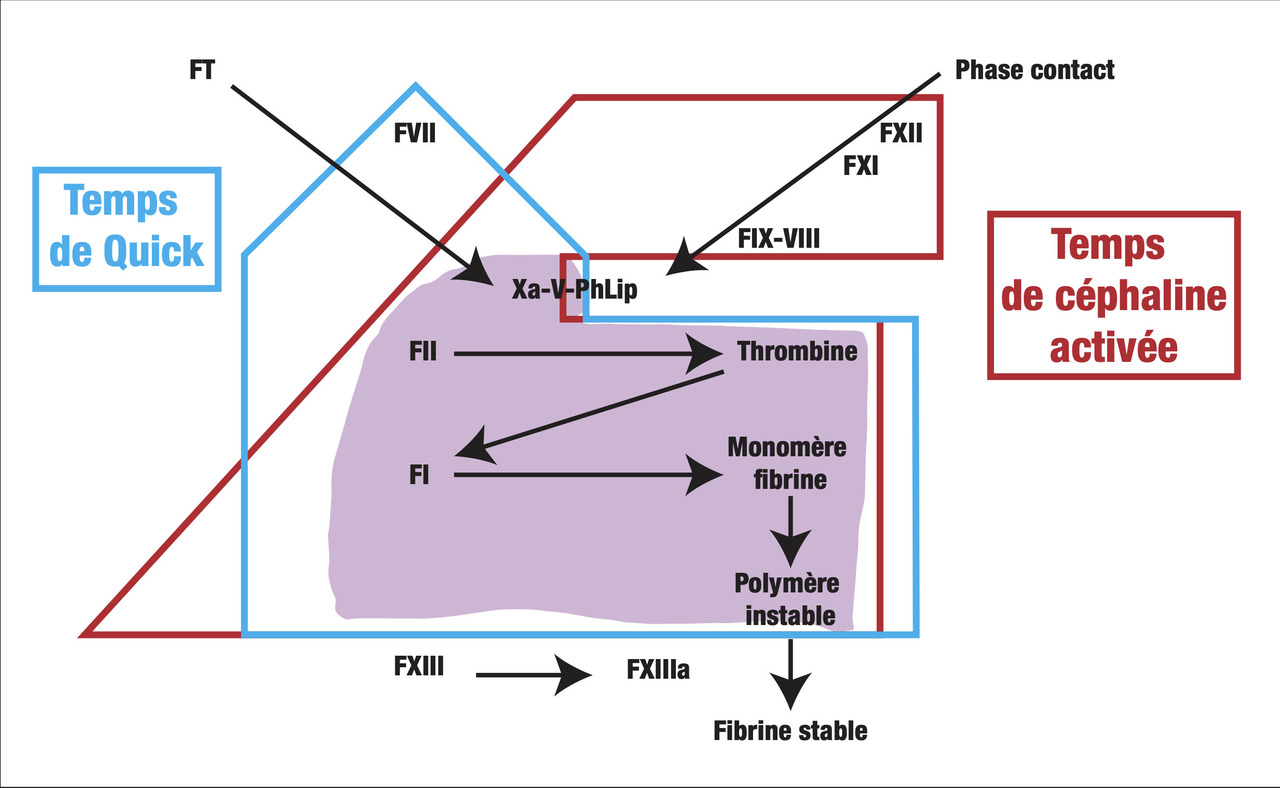
- le TCA ou temps de céphaline en présence d’activateur (seconde) : temps de coagulation d’un plasma citraté recalcifié en présence de phospholipides (qui remplacent les plaquettes) après activation complète de la phase contact de la coagulation. L’activateur utilisé peut être sous forme soluble (silice ou acide ellagique) ou sous forme de microparticules (kaolin) ;
- le TQ ou temps de Quick (seconde) : temps de coagulation d’un plasma citraté recalcifié en présence d’un excès de facteur tissulaire et de phospholipides procoagulants (thromboplastine) ;
- le TP ou temps de prothrombine (%) est l’expression du TQ du patient, rapporté à une droite d’étalonnage, obtenue par dilution d’un plasma normal. L’expression du TQ en INR (international normalized ratio) n’est utilisée que pour la surveillance des traitements par antivitamine-K ;
- le fibrinogène (g/L) : il s’agit d’une mesure fonctionnelle de l’activité coagulante, par un temps de coagulation du plasma dilué, en présence d’un excès de thrombine, qui est directement fonction du fibrinogène plasmatique.
Allongement isolé du temps de céphaline activée (
Il est important d’interroger le patient (ou son entourage) afin d’éliminer toute interaction médicamenteuse, notamment les héparines quel que soit leur type : héparine non fractionnée (HNF), héparine de bas poids moléculaire (HBPM) ou anticoagulants oraux directs (AOD).
Un mélange avec un plasma normal est parfois réalisé afin d’orienter le diagnostic : en cas de normalisation du TCA du patient par l’adjonction de plasma normal, on s’oriente vers un déficit factoriel ; l’absence de correction oriente vers la présence d’un anticoagulant circulant.
Rechercher les déficits de la voie plasmatique (= endogène = intrinsèque), soit les FVIII, FIX, FXI ; en cas de déficit, vérifier l’absence d’anticorps spécifique (anti-FVIII : hémophilie acquise ; anti-FIX ; anti-FXI) révélant un déficit acquis – en cas de déficit en FVIII toujours compléter par un dosage du FvW.
Phase contact, dosage de FXII et autres facteurs (kallicréine, pré-kallicréine, kininogène de haut poids moléculaire) : aucun de ces déficits n’est associé à un risque hémorragique.
Recherche de la présence d’un anticoagulant circulant de type lupique (ou antiprothrombinase) qui interfère sur les dosages ; ce type d’anticoagulant circulant est très fréquent chez le jeune enfant (notamment en cas d’infection ORL à répétition) ; il n’est pas associé à un risque hémorragique sauf en cas de spécificité contre un facteur plasmatique (par exemple anti-FII). Chez l’adulte, en dehors d’anticorps spécifique dirigé contre un facteur de coagulation, il doit orienter vers une pathologie dysimmunitaire (notamment le syndrome des antiphospholipides ou SAPL) et faire anticiper un risque thrombotique.
Diminution isolée du taux de prothrombine (TP) [équivalent normalisé de l’allongement du TQ] (
Rechercher un déficit des facteurs de la voie tissulaire (= voie extrinsèque = voie exogène) : facteur tissulaire (FT) et FVII. Le facteur tissulaire ne se dose pas.
En cas de déficit en FVII, vérifier l’absence d’anticorps spécifique.
S’assurer de l’absence de causes acquises comme les carences en vitamine K constitutionnelles ou acquises (AVK), les insuffisances hépatiques.
Allongement du temps de céphaline activée et diminution du taux de prothrombine (
Il faut avant tout éliminer un traitement par antivitamine-K (AVK) ou par les AOD (dabigatran, rivaroxaban, apixaban).
L’orientation se fait ensuite en dosant le fibrinogène :
- si le fibrinogène est normal : rechercher un déficit des facteurs de la voie commune, soit les FII, FV et FX. En cas de déficit d’un des facteurs, vérifier l’absence d’anticorps spécifique. Les déficits peuvent être isolés (d’origine constitutionnelle ou acquise) ou associés (le plus souvent acquis) : carence en vitamine K (diminution des FII, FVII, FX), insuffisance hépatique (baisse isolée ou non du FV) ;
- si le fibrinogène est abaissé : anomalie du fibrinogène quantitative (afibrinogénémie < 0,3 g/L ou hypofibrinogénémie entre 0,3 et 1 g/L) et/ou qualitative (dysfibrinogénémie ou dyshypofibrinogénémie).
Syndrome hémorragique clinique avec temps de céphaline activée, taux de prothrombine et fibrinogène normaux
Le seul facteur de coagulation non exploré par les bilans standard est le FXIII. Il est donc important, en cas de manifestations cliniques évocatrices (hémorragie du cordon, hémorragies intracrâniennes spontanées et à répétition, trouble de cicatrisation) et de bilan standard strictement normal, de demander spécifiquement le dosage de FXIII.
Fibrinolyse
Elle s’explore avant tout par le dosage des produits de dégradation du fibrinogène ou de la fibrine (PDF) ; les D-dimères sont spécifiques de la dégradation de la fibrine.
Certains centres ou services d’urgence et de réanimation utilisent le thrombo-élastogramme (TEG) ou thrombo-élastométrie rotative (ROTEM) afin d’évaluer la fibrinolyse en situation de choc hémorragique. Il s’agit de mesurer sur sang total la coagulation par enregistrement graphique des différentes phases de la coagulation et de la fibrinolyse.
Principales pathologies hémorragiques constitutionnelles ou acquises de l’hémostase et de la coagulation
Hémostase primaire
Les pathologies regroupent les thrombopénies (
Déficits en facteurs de von Willebrand
On en distingue 3 types : les déficits quantitatifs regroupant les types 1 (déficit partiel) et les types 3 (déficit complet) ; les déficits qualitatifs en FvW regroupés en types 2 variants (A, B, M, N…).
La forme modérée ou mineure (type 1) est la plus fréquente des pathologies hémorragiques constitutionnelles (
Le syndrome hémorragique est essentiellement cutanéo-muqueux : ecchymoses, purpura pétéchial, épistaxis, gingivorragies, ménométrorragies et hémorragies digestives. Il peut être spontané (forme les plus sévères) ou post-chirurgical (y compris après extraction dentaire).
Sur le plan biologique, on s’oriente le plus souvent par un allongement isolé du TCA (ou éventuellement du temps d’occlusion). La caractérisation du type se fait sur les dosages spécifiques du FvW (RCo et antigène) et du FVIII.
Il faut éliminer les diagnostics différentiels :
- l’hémophilie A ;
- le déficit acquis en FvW (facteur de von Willebrand acquis), survenant en l’absence de contexte familial et le plus souvent associé à une autre pathologie (hypothyroïdie, gammapathie monoclonale [IgM], rétrécissement aortique, thrombocytémie essentielle).
Les principes thérapeutiques reposent sur :
- l’administration de desmopressine ou DDAVP ou hormone antidiurétique (dans les types 1 ou certains types 2) qui relarguent les réserves endothéliales chez les bons répondeurs. Cette administration peut être réalisée par voie intraveineuse ou intranasale toutes les 12 ou 24 heures. Elle permet une augmentation rapide (environ 30 min) du taux de FvW. Cette réponse doit être évaluée chez les patients par un test préthérapeutique, et son utilisation ne peut pas dépasser 3 à 4 injections successives en raison d’un phénomène d’épuisement (tachyphylaxie). Il est indispensable en raison de la nature de la molécule d’y associer une restriction hydrique pendant 12 heures après chaque utilisation ;
- en l’absence de réponse au DDAVP ou dans les formes sévères, on utilise des concentrés de FvW purifiés par voie intraveineuse ;
- pour la prise en charge des saignements muqueux modérés, les antifibrinolytiques peuvent être associés ;
- ne pas oublier : la contre-indication des médicaments, antiagrégant plaquettaire ou anticoagulant, sans avis spécialisé ; la contre-indication des injections intramusculaires ; l’avis spécialisé indispensable avant tout geste invasif ou chirurgical.
Les thrombopathies (constitutionnelles ou acquises)
Dans la majorité des cas, il s’agit de pathologies acquises secondaires à une prise médicamenteuse ou une pathologie chronique associée (insuffisance rénale, hépatique, syndrome myéloprolifératif). Les thrombopathies constitutionnelles sont évoquées le plus souvent chez l’enfant et en présence d’antécédents familiaux hémorragiques. Elles restent exceptionnelles et résumées dans le
Les signes cliniques sont principalement cutanéo-muqueux, spontanés et d’importance variable selon l’étiologie : purpuras, ecchymoses, gingivorragies, épistaxis, ménométrorragies, hémorragies peropératoires ou post-chirurgicales immédiates.
Le diagnostic se fait sur des tests spécifiques (agrégations et cytométrie plaquettaire) en centre spécialisé.
La prise en charge et le traitement nécessitent un avis spécialisé.
Coagulation
Les pathologies de la coagulation regroupent les déficits constitutionnels ou acquis en facteur coagulant.
La prévalence des différents déficits en facteur coagulant est résumée dans le
Hémophilies
C’est la plus fréquente des pathologies hémorragiques sévères. On distingue deux types d’hémophilie : l’hémophilie A secondaire à un déficit en FVIII et l’hémophilie B secondaire à un déficit en FIX. C’est une maladie génétique (transmission récessive liée à l’X) rare. La maladie s’exprime principalement chez les garçons du fait de sa transmission, mais les femmes peuvent être symptomatiques en raison d’un taux abaissé de facteur (« conductrice à taux bas » ; lyonisation de l’X).
L’importance des manifestations hémorragiques est proportionnelle à la sévérité du déficit en FVIII ou FIX. Pour les 2 types d’hémophilie, il existe 3 formes : sévère pour un déficit complet < 1 % de facteur, modérée pour un déficit compris entre 1 et 5 % et mineure pour un déficit compris entre 5 % et 40 %.
Dans les formes sévères, en l’absence d’antécédent familial connu, les premières manifestations surviennent généralement à l’âge de la marche. Les principaux saignements sont les atteintes articulaires (hémarthroses) ou les hématomes profonds (musculaires ou plus rarement viscéraux) ; ils surviennent spontanément ou pour des traumatismes minimes. Dans le cas de formes mineures, le diagnostic a lieu le plus souvent de manière fortuite sur un bilan préopératoire ou sur une complication hémorragique (saignements prolongés, hématome) après un geste invasif (extraction dentaire) ou une chirurgie.
Le diagnostic biologique se fait sur un allongement isolé du TCA (TQ, TP et fibrinogène normaux) et sur le dosage spécifique des facteurs VIII et IX.
Dans l’hémophilie A, le principal diagnostic différentiel des formes modérées ou mineures est le déficit en FvW, et il faut donc toujours associer un dosage spécifique du FvW. Chez les adultes, il faut également éliminer une hémophilie acquise (anticorps spécifique anti-FVIII). Ce type de pathologie se voit essentiellement chez le sujet âgé (syndrome paranéoplasique, syndrome myéloprolifératif, gammapathie, dysimmunité) ou chez la femme jeune en période gravidique.
Dans l’hémophilie B mineure, il convient d’éliminer une diminution des autres facteurs vitamine K-dépendant, afin d’éliminer une cause acquise.
La prise en charge s’effectue par les centres de traitement des hémophiles (il en existe plus de 30 en France métropolitaine). Le traitement substitutif peut être administré à la demande (en cas d’accident hémorragique ou de geste invasif ponctuel) ou dans le cadre d’une prophylaxie (prévention des saignements intra-articulaires, prévention pour des interventions chirurgicales). Il s’agit de concentrés purifiés de FVIII (hémophilie A) ou FIX (hémophilie B) d’origine plasmatique (fractionnement de plasma) ou recombinant. Ces produits sont classés parmi les médicaments dérivés du sang et sont soumis à une traçabilité. Ils doivent être administrés par voie intraveineuse stricte.
Les patients sont porteurs en permanence d’une carte nationale comportant les caractéristiques de l’hémophilie, les particularités ou complications, la conduite à tenir en cas d’urgence et les coordonnées du centre spécialisé (centre de traitement des hémophiles).
Le traitement substitutif expose principalement à une complication majeure : l’apparition d’un inhibiteur (anticorps dirigé contre le facteur déficitaire), rendant le traitement habituel inefficace et obligeant le recours à des molécules by-passant (court-circuitant la voie de la coagulation habituelle du FVIII ou FIX) : complexe prothrombinique ou FVII activé. Ce risque concerne surtout l’hémophilie A avec une fréquence d’environ 20 %. Même si les progrès ont été majeurs concernant la sécurisation des médicaments dérivés du sang, le risque lié à la transmission possible d’agent infectieux reste une préoccupation des médecins et des patients.
Dans le cas de l’hémophilie A mineure (uniquement), comme pour les déficits en FvW, la desmopressine (DDAVP ou Minirin) peut être utilisée chez ces patients après avoir démontré son efficacité par un test thérapeutique.
Toute prise en charge d’un hémophile en urgence ou tout geste invasif (même minime) nécessite une prise de contact avec le centre de traitement du patient afin de discuter du traitement adapté à la situation, et ce quelle que soit la sévérité de l’hémophilie.
Hypovitaminose K et insuffisance hépatocellullaire
Hypovitaminose K
Les principales sources de vitamine K sont l’alimentation et la synthèse intestinale par la flore microbienne. Elle est solubilisée par les sels biliaires puis absorbée par l’intestin grêle. C’est le cofacteur d’une carboxylase hépatocytaire qui intervient pour l’activation des facteurs de coagulation vitamine K-dépendants synthétisés par le foie (FII, FVII, FIX, FX). Une carence entraîne alors une baisse de synthèse de ces facteurs et peut conduire à un syndrome hémorragique.
Les causes carentielles sont différentes chez le nouveau-né, l’enfant et l’adulte.
En pédiatrie, elles touchent principalement le nouveau-né qui n’a aucun stock vitaminique, pas de flore microbienne et un apport alimentaire pauvre. Jusqu’à la substitution systématique en vitamine K1, elle était la principale cause de la maladie hémorragique du nouveau-né survenant entre 2 et 6 jours. La supplémentation doit se poursuivre tant qu’il existe un allaitement maternel exclusif car la concentration en vitamine K est faible (quelle que soit l’alimentation).
Chez l’enfant et l’adulte, ces carences sont secondaires :
- post-médicamenteuse : traitement antivitamine-K (AVK) ou antibiothérapie prolongée (destruction de la flore microbienne) ; absorption de produit bloquant la synthèse de vitamine K (produits ou dérivés raticides) qui peut être volontaire (autolyse) ou accidentelle (empoisonnement) ;
- carences d’apport : rares en dehors des malnutritions sévères, de l’anorexie ou d’alimentations parentérales prolongées ;
- troubles d’absorption intestinale : par défaut de solubilisation (pathologie des voies biliaires) ou en raison d’une pathologie intestinale chronique (maladie cœliaque, résection intestinale, colite ulcéreuse).
Le diagnostic est évoqué sur l’allongement du TCA et du TQ (baisse du TP) secondaire à une diminution des facteurs vitamine K dépendants (FII, FVII, FIX et FX), alors que le FV et le fibrinogène sont normaux, témoignant de l’absence d’atteinte hépatique.
Le traitement consiste à apporter de la vitamine K1 per os, par voie IV ou IM en fonction de la cause initiale. Il est important de prévenir ces carences chez les patients à risque (nouveau-né : 2 mg per os à J1, nutrition parentérale : apport IV). La correction des facteurs se fait en 6-12 heures. En cas de syndrome hémorragique, l’apport per os peut suffire (enfant : 1 à 2 mg per os ou IV ; adulte : 10-20 mg per os ou IV) ; si le saignement est sévère, l’apport de concentrés de facteurs du complexe prothrombique (Kanokad ou Octaplex, par exemple) est indispensable pour une action rapide.
L’insuffisance hépatocellulaire
Le foie est le lieu principal de synthèse des facteurs de l’hémostase (procoagulant ou inhibiteurs). Les anomalies de l’hémostase qui en découlent dépendent avant tout de la sévérité de la pathologie hépatique.
Elle est évoquée comme précédemment sur l’association d’un allongement du TCA et du TQ (baisse du TP) avec une atteinte de tous les facteurs synthétisés par le foie : facteur de la coagulation vitamine K-dépendant (FII, FVII, FIX, FX) ou non (FV, fibrinogène) mais également des protéines inhibitrices (antithrombine [AT], protéines C et S). Seul le FVIII reste normal (voire augmenté) car sa synthèse a lieu également dans les cellules endothéliales.
Lors des atteintes sévères, il existe le plus souvent une thrombopénie associée (séquestration splénique). En cas d’hépatite aiguë, c’est le FV qui sert de facteur pronostique.
D’autres atteintes de l’hémostase peuvent être observées : hyperfibrinolyse, thrombopathie acquise.
La prise en charge dépend principalement du traitement de l’étiologie de l’insuffisance hépatique. En cas de syndrome hémorragique secondaire à un déficit en facteur de coagulation, l’apport de plasma frais congelé sécurisé est le plus adéquat. L’apport de vitamine K est inutile. En cas de fibrinolyse, il faut associer des antifibrinolytiques (acide tranexamique).
Coagulation intravasculaire disséminée (CIVD)
Il s’agit d’une activation systémique et excessive de la coagulation. Elle peut être localisée (malformation artério-veineuse, angiomes) ou le plus souvent généralisée (choc septique). Elle peut apparaître sous une forme aiguë (plus fréquente) ou chronique (malformation ou néoplasie). Elle associe des manifestations cliniques et biologiques variables. Elle peut être déclenchée par différents mécanismes :
- l’exposition non contrôlée de facteur tissulaire (lésion d’organe riche en facteur tissulaire : placenta, prostate, utérus, poumon) ;
- l’expression pathologique du facteur tissulaire, par les cellules endothéliales ou les monocytes, induite par l’endotoxine, les cytokines et complexes antigène-anticorps ; l’expression d’un analogue du facteur tissulaire par des cellules tumorales (LAM3, cancer du poumon, du pancréas ou de la prostate) ;
- la présence anormale d’enzymes protéolytiques en circulation comme lors d’une morsure de serpent (venin) ou lors des pancréatites aiguës (trypsine).
Cette activation anormale de la coagulation va aboutir à une production de thrombine (FII) en excès, entraînant la transformation du fibrinogène en fibrine et l’activation des plaquettes qui va aboutir à la formation de micro-thrombi dans la circulation. Il s’ensuit une consommation des facteurs de la coagulation, des plaquettes entraînant un risque hémorragique.
Les principales causes sont regroupées dans le
Le diagnostic est évoqué devant un tableau clinique évocateur dans les circonstances citées.
La coagulation intravasculaire disséminée aiguë se présente fréquemment par un tableau clinique thrombotique et hémorragique souvent « catastrophique » avec des microthromboses diffuses responsables de nécroses organiques ischémiques (peau, poumon, rein, cerveau…), des hémorragies provoquées (post-geste invasif : ponction veineuse ou artérielle, ou chirurgie) ou spontanées (cutanéo-muqueuses), associé à des signes d’hypovolémie/hypotension, voire de choc hémorragique.
Les formes chroniques sont souvent associées aux pathologies malignes ou aux malformations vasculaires, et se présentent sous la forme d’un tableau prothrombotique prédominant où le syndrome hémorragique est souvent au second plan ou mineur.
Le diagnostic biologique est parfois délicat dans les formes aiguës de coagulation intravasculaire disséminée. On observe un allongement du TQ (baisse TP) secondaire à la diminution des FII, FVII, FIX, FX puis du FV. Le fibrinogène diminue. Une thrombopénie s’associe rapidement au tableau. C’est la cinétique de ces diminutions entre deux prélèvements qui a une valeur diagnostique plus que le chiffre observé.
Le dosage des D-dimères est essentiel. Leur présence témoigne de l’action directe de la plasmine sur la fibrine. En cas de négativité, ceci exclut le diagnostic de coagulation intravasculaire disséminée. Au contraire, leur positivité n’est pas spécifique de la coagulation intravasculaire disséminée car ils augmentent dans diverses situations (thrombose, embolie, infection). Les complexes solubles (CS) sont des complexes formés entre les monomères de fibrine et les produits de dégradation de la fibrine ; attention ! en cas de fibrinogène très bas, ces complexes ne peuvent se former, leur négativité n’élimine pas le diagnostic.
Il existe un score diagnostique de coagulation intravasculaire disséminée (
Le principal diagnostic différentiel est la fibrinolyse primitive. Il s’agit d’un phénomène rare lié à la libération massive de l’activateur tissulaire du plasminogène (tPA) secondaire à une chirurgie hépatique, vasculaire ou pulmonaire, ou en cas de néoplasie (prostate) ou de pathologie obstétricale. Dans ce cas, le tableau est uniquement hémorragique. Les signes biologiques sont proches, avec une hypofibrinogénémie, mais il n’y a ni CS ni D-dimères élevés.
La prise en charge dépend de la cause car c’est le traitement de l’étiologie principale qui permettra de traiter la coagulation intravasculaire disséminée. Le traitement substitutif en parallèle correspond à un support transfusionnel adapté (concentrés plaquettaires et plasma frais congelé sécurisé ou viro-atténué). Les concentrés spécifiques de facteur de coagulation sont habituellement contre-indiqués en dehors de l’apport éventuel de fibrinogène en cas d’hypofibrinogénémie sévère et de syndrome hémorragique majeur. Les concentrés d’antithrombine et de protéine C n’ont pas fait la preuve de leur efficacité. L’héparine reste parfois utilisée dans le versant thrombotique.
POINTS FORTS À RETENIR
L’interrogatoire et l’examen clinique sont indispensables à l’interprétation d’un bilan de coagulation.
Le bilan de coagulation ne s’interprète pas sans le dosage du fibrinogène.
Tout allongement du temps de céphaline activée nécessite le dosage des FVIII, FIX, FXI et la recherche d’un anticoagulant circulant.
Tout allongement du temps de Quick (ou baisse du TP) nécessite le dosage des FII, FV, FVII et FX.
Devant un syndrome hémorragique clinique, la liste des médicaments consommés doit être exhaustive.
Les déficits en facteur de la coagulation sont des maladies rares.
La présence d’un anticoagulant circulant chez l’enfant de moins de 6 ans est fréquent et sans risque hémorragique quel que soit l’allongement du temps de céphaline activée.
Schveb JF. Maladies hémorragiques constitutionnelles. Rev Prat 2015;65: à paraître.
Samama MM et al. Collection Abrégé MASSON: Hémorragies et thrombose - du diagnostic au traitement.
Sampol J, Arnoux D, BoutièreB. Collection Option Bio - Elsevier: Manuel d'hémostase
Rubio MT, Dargaud Y, Ghesquières H, Fayette J. Collection ECN Med - Editions Pradel: Hématologie Candérologie.
Lehot JJ, Ricaud X. Cas clinique pour ECN. Editions Pradel: Cancérologie Oncohématologie Hématologie.
 Encadrés
Encadrés