Savoir diagnostiquer et connaître les principes du traitement d’une acidose métabolique, d’une acidose ventilatoire, d’une hypokaliémie, d’une hyperkaliémie, d’une hyponatrémie, d’une hypernatrémie, d’une hypocalcémie.
Principes de physiologie rénale
Pour comprendre les dysnatrémies, il est essentiel de connaître quelques principes de physiologie rénale.
Un composé osmotiquement actif est un soluté qui entraîne un mouvement d’eau de part et d’autre d’une membrane cellulaire. L’osmolarité s’équilibre entre les compartiments intra- et extracellulaires, soit de part et d’autre de la membrane cellulaire. Cet équilibre sous-tend le volume cellulaire. De façon physiologique, la quantité intracellulaire d’osmoles est fixe, son changement de concentration (soit le changement de l’osmolarité) dépend donc le plus souvent d’un excès ou d’un manque d’eau pure. La concentration des osmoles peut être exprimée par l’osmolalité (quantité d’osmoles par kg d’eau – véritable reflet du pouvoir osmotique) ou par l’osmolarité (quantité d’osmoles par litre de plasma). En pratique, on parle plus souvent d’osmolarité, mais ces deux paramètres sont équivalents. Une hyperosmolarité plasmatique correspond donc à une déshydratation intracellulaire et une hypo-osmolarité plasmatique à une hyperhydratation intracellulaire.
L’osmolarité normale est de 300 mOsm/kg d’eau. L’osmolarité peut être mesurée ou estimée par la formule suivante (qui tient compte de la natrémie, de la kaliémie et de la glycémie, qui sont les substances osmotiques principales à l’état physiologique) : Osm = (Na+ + K+) × 2 + glycémie.
L’osmole majoritaire au niveau extracellulaire est le sodium (Na+), et celle majoritaire au niveau intracellulaire est le potassium (K+). Ainsi, la mesure de la concentration en sodium extracellulaire (la natrémie) reflète l’hydratation intracellulaire dans un grand nombre de cas, sauf en cas d’erreur de mesure (fausse hyponatrémie dans le cas d’hyperprotidémie ou d’hyperlipidémie majeure) ou en cas de présence d’une osmole supplémentaire anormale (hyperglycémie, mannitol, etc.). En cas de suspicion de dysnatrémie, il est souhaitable, autant que possible, de mesurer l’osmolalité plasmatique (sur automate, le plus fiable), plutôt que de l’estimer/la calculer à partir de la natrémie. L’eau constitue environ 60 % du poids du corps d’un homme adulte jeune. Cette composition varie avec l’âge et le sexe. La majorité de l’eau est contenue dans le compartiment intracellulaire (deux tiers du stock hydrique). Le tiers restant constitue le volume extracellulaire, réparti pour trois quarts dans le compartiment interstitiel et un quart dans le compartiment vasculaire. Ainsi, les différents volumes de répartition de l’eau peuvent être répartis en douzièmes : 1/12e en intravasculaire, 3/12es en interstitiel, 8/12es en intracellulaire.
Il est fondamental de distinguer la régulation du bilan de l’eau (osmolarité/osmolalité ou hydratation intracellulaire) et celle du bilan du sodium (volémie ou hydratation extracellulaire). Le bilan hydrique dépend des apports (le volume hydrique absorbé) et des pertes (transpiration, pertes digestives et surtout volume urinaire). Le volume urinaire est la variable d’ajustement, régulé par l’hormone antidiurétique (ADH) produite par l’hypothalamus et stockée dans la post-hypophyse, et qui stimule l’expression d’aquaporines permettant la réabsorption d’eau libre dans le canal collecteur rénal. La régulation du sodium détermine la volémie, modulée au niveau rénal par le système rénine-angiotensine-aldostérone (SRAA) qui permet la stimulation de la réabsorption de sodium par le canal ENaC (au niveau du canal collecteur). Les reins aident au maintien de la volémie et de l’équilibre hydrique en régulant à la fois la quantité de sodium et d’eau libre dans les urines.
Concernant la capacité de dilution et de concentration de l’urine, les reins ne peuvent pas éliminer de l’eau pure, il existe une dilution maximale des urines. Cela signifie que, pour éliminer de l’eau, il nous faut un minimum d’apports d’osmoles. En pratique, si l’on imagine que nos reins ont une capacité de dilution « bloquée » à 500 mOsm/L, cela signifie que pour éliminer 1 litre d’eau, il faut éliminer 500 mOsm (provenant du catabolisme des protéines endogènes et des apports). Cette capacité de dilution et de concentration varie, pour une personne jeune avec des reins fonctionnels, de 60 mOsm/kg à 1 200 mOsm/kg.
L’hormone antidiurétique (ADH ou vasopressine) permet de réguler le bilan hydrique. Il existe deux stimuli principaux à la sécrétion d’ADH : l’hyperosmolalité cellulaire et la volémie. Le stimulus volémique est plus fort que le stimulus osmotique. Ainsi, en cas d’hypovolémie, la sécrétion d’ADH se poursuit malgré la possible apparition d’une hyponatrémie. C’est la quantité d’ADH sécrétée qui fait varier la concentration osmolaire des urines. Si le taux d’ADH est effondré et que la fonction rénale est normale, l’osmolalité urinaire est inférieure à 100 mOsm/L. Si la sécrétion d’ADH est maximale, comme dans une situation de grande déshydratation globale, l’osmolalité urinaire est au maximum, soit 1 200 mOsm/L.
Hyponatrémie
Définition (rang A)
L’hyponatrémie est définie par une natrémie inférieure à 135 mmol/L (sévère si ≤ à 120 mmol/L) et reflète une hyperhydratation intracellulaire (en l’absence d’erreur de mesure ou d’une substance osmotiquement active anormalement présente). Ainsi, une hyponatrémie « vraie » s’accompagne d’une osmolalité diminuée, inférieure à 275 mOsm/L. La présence d’une substance osmotiquement active supplémentaire peut donner une hyponatrémie hyperosmolaire (Osm > 300 mOsm/L) tandis qu’une anomalie de mesure (liée à une hyperlipidémie ou une hyperprotidémie majeure) peut donner une hyponatrémie iso-osmolaire. Dans ces deux dernières situations, l’hyponatrémie ne reflète donc pas obligatoirement une hyperhydratation intracellulaire.
Bilan étiologique (rang B)
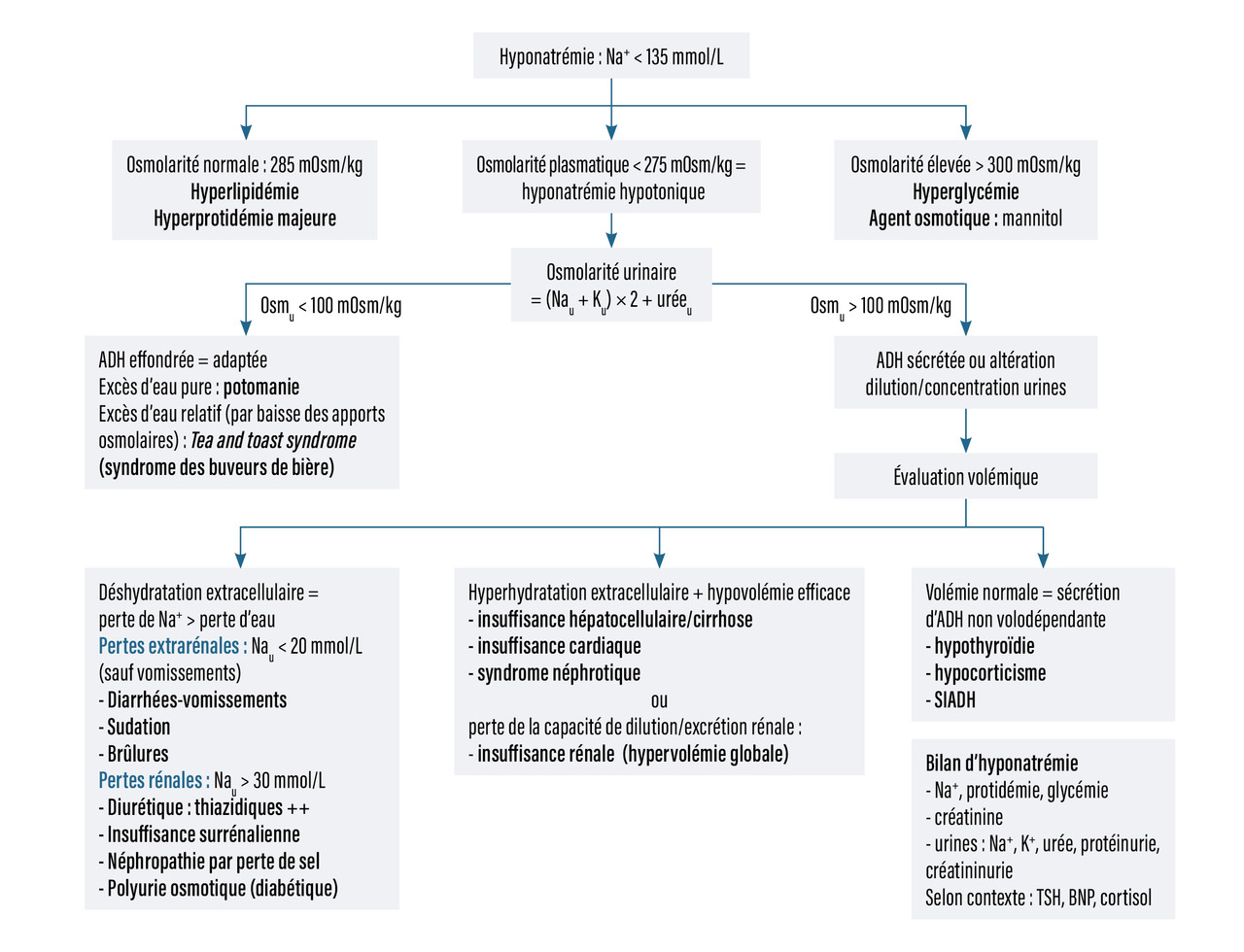
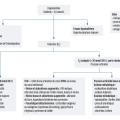
La démarche diagnostique de l’hyponatrémie est résumée dans la figure 1.
La première étape consiste à s’assurer qu’il s’agit bien d’une hyponatrémie vraie, c’est-à-dire une hyponatrémie hypo-osmolaire, et donc à éliminer les causes d’hyponatrémie hyper- ou iso-osmolaire (voir plus haut).
La seconde étape est de calculer l’osmolarité urinaire, par la formule : Osmolarité urinaire = (Nau + Ku) × 2 + urée u.
Dans les causes d’hyponatrémie vraie avec une osmolarité urinaire effondrée, les reins fonctionnent parfaitement, l’ADH est effondrée et les urines très diluées. L’hypo-osmolalité survient car la capacité de dilution maximale des urines est dépassée : l’apport d’eau est relativement trop important par rapport à l’apport osmolaire pour que l’eau soit excrétée. Par exemple, si une personne absorbe 4 L d’eau et uniquement 100 osmoles sur vingt-quatre heures, avec une capacité de dilution maximale de 100 mOsm/L, les reins n’arriveront à éliminer (en diluant au maximum les urines) que 1 L d’eau, le gain sera donc de 3 L net d’eau.
Lorsqu’il existe une hyponatrémie vraie avec une osmolarité urinaire supérieure à 100 mOsm/L, cela signe une sécrétion persistante d’ADH (ou une altération de la capacité de dilution et de concentration urinaire) qui n’est donc pas adaptée à cette hypo-osmolalité. Il faut alors évaluer la volémie pour identifier trois situations principales : une sécrétion d’ADH volodépendante secondaire à une hypovolémie vraie (déshydratation extracellulaire), une sécrétion volodépendante secondaire à une hypovolémie efficace (baisse de perfusion des récepteurs artériels, qui peut être accompagnée d’une inflation veineuse et/ou interstitielle et donc d’un volume extracellulaire augmenté), ou une sécrétion inappropriée d’ADH. Les situations correspondantes sont détaillées dans la figure 1 (en pratique clinique, les diurétiques thiazidiques peuvent fréquemment entraîner une hyponatrémie avec une volémie peu diminuée. Cela provient probablement de mécanismes plus complexes et d’une prédisposition génétique).
Lors d’une insuffisance rénale chronique avancée, il existe une perte de la capacité de dilution et de concentration urinaire, pouvant être responsable d’une hyperhydratation globale.
Le syndrome de sécrétion inappropriée de l’ADH (SIADH) correspond à la situation au cours de laquelle l’ADH est sécrétée sans stimulus volémique ou osmotique. On observe alors une hyponatrémie vraie chez un patient cliniquement euvolémique. Deux endocrinopathies peuvent mimer ce tableau : l’hypothyroïdie et l’hypocorticisme. Les causes du SIADH sont nombreuses, mais les plus fréquentes sont médicamenteuses (antipsychotiques, notamment carbamazépine, inhibiteurs de la recapture de la sérotonine…) et paranéoplasiques (cancer du poumon, lymphome…).
Retentissement clinico-biologique (rang B)
Il est non spécifique. On distingue habituellement les signes modérément sévères (nausées, confusion, céphalées) et sévères (vomissements, somnolence parfois jusqu’au coma profond, épilepsie, détresse cardiorespiratoire).
Traitement (rang C)
Il varie en fonction de la cause et de la sévérité, mais la restriction hydrique est toujours efficace. En cas d’hyponatrémie chronique, le rythme de correction de la natrémie ne doit pas dépasser 10 mmol/L les vingt-quatre premières heures et 18 mmol/L au total les quarante-huit premières heures. Il existe un risque neurologique lié à la correction trop rapide de l’hyponatrémie (encadré 1).
Les principes du traitement selon la cause sont les suivants :
- potomanie : traitement de la pathologie psychiatrique pour diminuer la prise hydrique ;
- tea and toast syndrome (syndrome des buveurs de bière) : diminuer la prise hydrique et augmenter les apports osmolaires ;
- hypovolémie vraie : rétablir la volémie par hydratation avec eau et osmoles, en milieu hospitalier par des solutés contenant du sodium comme le NaCl 0,9 %, le Ringer Lactate, le bicarbonate de sodium 1,4 %. Si réhydratation à domicile, on peut proposer de l’eau classique en y associant des apports alimentaires non limités en sel et/ou de l’eau contenant beaucoup d’osmoles, comme l’eau de Vichy ;
- en cas d’hypovolémie efficace associée à une hyperhydratation extracellulaire, le traitement de la cause (si possible) est associé aux diurétiques et à une restriction hydrique ;
- en cas d’hypervolémie globale, la prise en charge repose sur les diurétiques et la restriction hydrique ;
- en cas de SIADH, la mesure thérapeutique essentielle est la restriction hydrique. En l’absence d'amélioration, les apports osmolaires peuvent être majorés (notamment par l’utilisation de sachets d’urée), de même que la perte hydrique, grâce à des médicaments aquarétiques (inhibiteurs de la vasopressine, sur avis spécialisé).
En cas de symptômes sévères (voir plus haut), un traitement d’urgence par administration de sérum salé hypertonique (NaCl 3 %) est à introduire en milieu hospitalier, associé aux anticonvulsivants en cas de crise convulsive. Ce traitement permet de corriger rapidement la natrémie (dans la limite de 6 mmol/L les six premières heures), permettant la plupart du temps la correction du symptôme, tout en ne dépassant pas le rythme de correction quotidien recommandé.
Hypernatrémie (rang C)
Définition
L’hypernatrémie est définie par une natrémie supérieure à 145 mmol/L et reflète une déshydratation intracellulaire. L’osmolarité est alors supérieure à 300 mOsm/L.
Il n’existe pas de fausses hypernatrémies, l’hypernatrémie est toujours témoin d’une hyperosmolarité plasmatique et d’une déshydratation intracellulaire.
Bilan étiologique
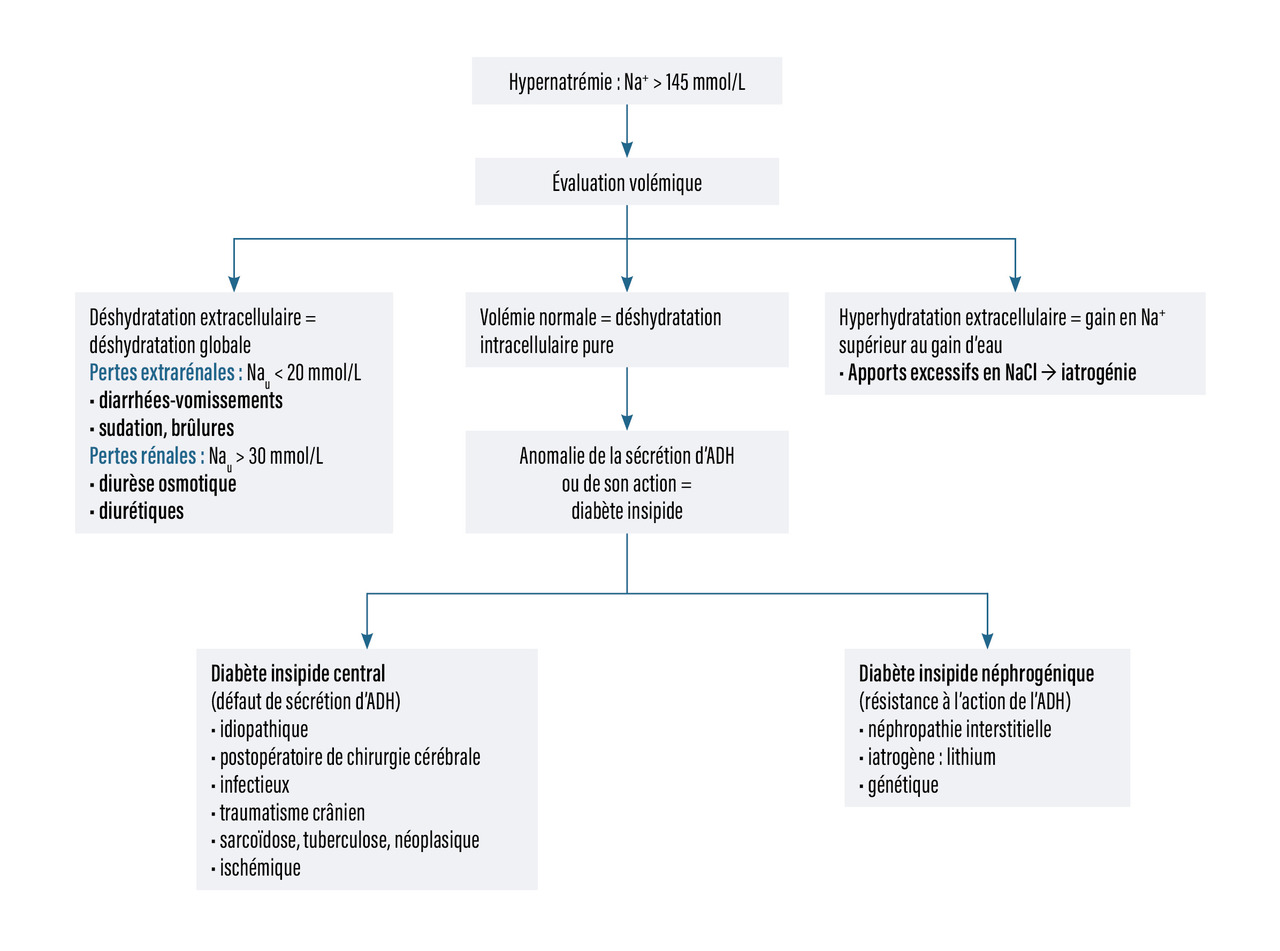
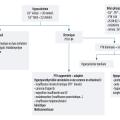
La démarche diagnostique de l’hypernatrémie est résumée dans la fig. 2.
L’hypernatrémie/hyperosmolalité plasmatique signe systématiquement une déshydratation intracellulaire. Ce trouble ne survient que lorsque l’accès à l’eau est limité. S’il n’existe pas de fausse hypernatrémie, dans certains cas, la natrémie peut toutefois sous-estimer l’hyperosmolalité ; c’est par exemple le cas dans le cadre du coma hyperosmolaire dans le diabète, où la glycémie élevée a un pouvoir osmotique important.
Retentissement clinico-biologique
Le principal signe d’hypernatrémie est la soif, qui peut être intense. La sécheresse des muqueuses et la présence d’un pli cutané sont d’une sensibilité et d’une spécificité faibles. La présence de signes neurologiques signe une hypernatrémie grave : confusion, somnolence, convulsion, coma, etc.
Traitement
La correction rapide d’une hypernatrémie chronique chez l’adulte ne présente pas le même risque que la correction rapide d’une hyponatrémie. Il reste néanmoins recommandé de ne pas dépasser un rythme de correction supérieure ou égale à 10 mmol/L par vingt-quatre heures.
Le traitement consiste en la correction du déficit hydrique par des apports d’eau orale et/ou avec une perfusion intraveineuse de soluté ne contenant pas de sodium (glucosé 2,5 % ou 5 %).
Dans le cas d’une déshydratation globale, il s’agit à la fois de corriger le déficit en sel et en eau, donc d’apporter des solutés isotoniques ainsi que de l’eau.
Dans le cas d’une hyperhydratation extracellulaire associée, la cause la plus fréquente est l’hypernatrémie iatrogène en hospitalisation. Il faut alors, selon la situation, diminuer l’apport en NaCl ou augmenter l’apport en eau pure.
En cas de diabète insipide, l’hypernatrémie n’apparaît que dans les situations où il existe une baisse des apports hydriques. Dans les causes néphrogéniques, la prescription d’apports hydriques importants (parfois plusieurs litres d’eau par jour) constitue le seul traitement. Lorsqu’un traitement par lithium est à l’origine du diabète insipide, sa poursuite doit être discutée avec le psychiatre en fonction du rapport bénéfice-risque et de la capacité du patient à poursuivre l’hyperhydratation.
Dans les diabètes insipides d’origine centrale, l’apport d’ADH exogène peut se discuter.
Dyskaliémie
Principes de physiologie rénale (rang B)
Le potassium est le principal cation du milieu intracellulaire (concentration intracellulaire entre 100 et 150 mEq/L) : la kaliémie représente une faible proportion du stock total mais est très finement régulée, avec des valeurs normales entre 3,5 et 5 mmol/L. Plusieurs acteurs interviennent dans cette régulation. La toxicité cardiaque signe la gravité d’une dyskaliémie. L’hyperkaliémie induit une hypoexcitabilité cardiaque et l’hypokaliémie une hyperexcitabilité cardiaque.
Les dyskaliémies surviennent par le biais de différents mécanismes.
Apports en potassium : les carences d’apports sont exceptionnelles et concernent essentiellement les grandes dénutritions et les anorexies mentales (dans lesquelles peut aussi s’associer une augmentation des pertes par la prise de diurétique/laxatif). En situation physiologique (hors insuffisance rénale avancée et iatrogénie médicamenteuse), une augmentation des apports alimentaires n’entraîne pas d’hyperkaliémie.
La pompe Na-K-ATPase est ubiquitaire et constitue le principal régulateur du transfert intracellulaire de potassium ; elle est à l’origine de la création d’un potentiel de repos transmembranaire (différence de répartition naturelle du sodium et du potassium intra-/extracellulaire induisant une polarisation membranaire). Son activation (par les catécholamines ou l’insuline, par exemple) stimule l’entrée du potassium du compartiment extracellulaire vers le compartiment intracellulaire (diminue la kaliémie) ; elle permet l’entrée de deux ions K+ en échange de trois ions Na+. Au contraire, son inhibition (acidose métabolique, bêtabloquants non sélectifs, intoxication digitalique) entraîne une augmentation de la kaliémie.
Le rein est le principal acteur de la régulation de la kaliémie. L’insuffisance rénale aiguë (IRA) peut s’accompagner d’une hyperkaliémie par baisse de l’excrétion rénale et par transfert du secteur intracellulaire vers l’extracellulaire, liée à l’acidose associée. L’hyperkaliémie sévère dans le contexte de l’IRA anurique est une urgence nécessitant une dialyse. Dans l’insuffisance rénale chronique (IRC) non terminale, l’hyperkaliémie est le plus souvent modérée, mais les patients deviennent plus sensibles aux apports élevés de potassium.
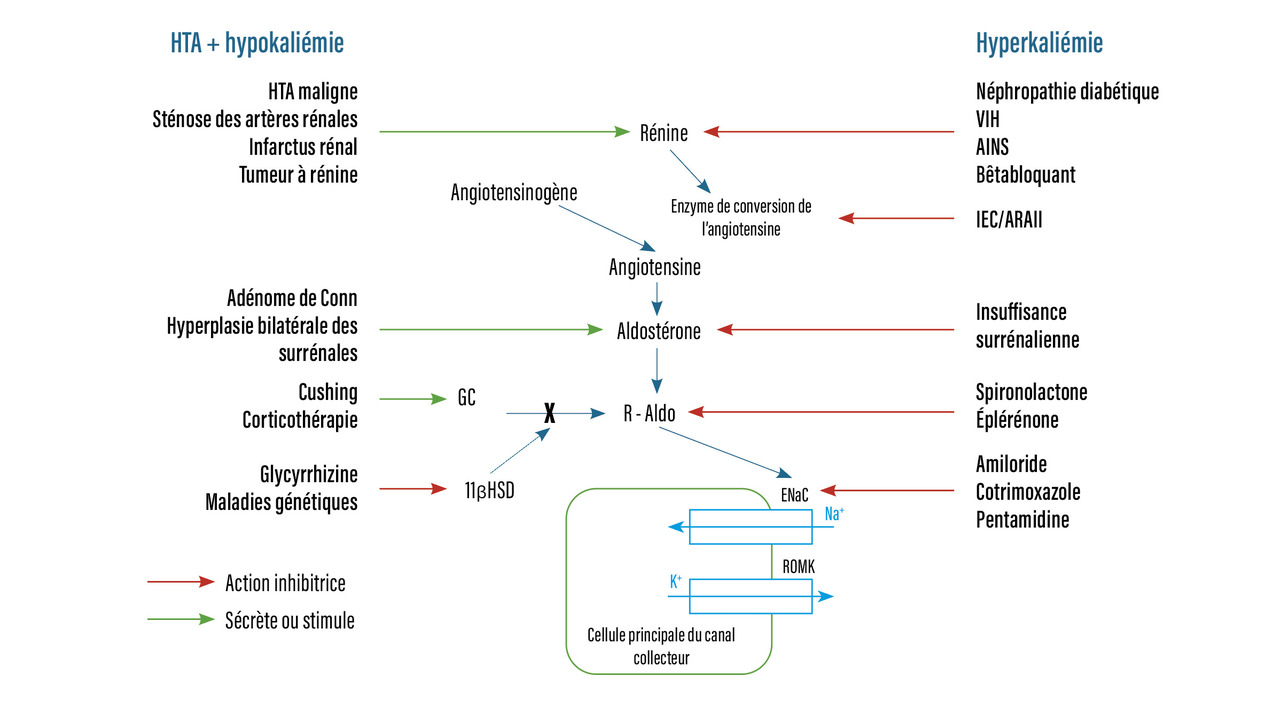
La cellule principale du canal collecteur rénal, et particulièrement le canal ROMK (stimulé par l’aldostérone), est le principal régulateur de l’excrétion potassique au niveau rénal. À ce niveau du tubule rénal, l’aldostérone, en se liant à son récepteur, stimule la réabsorption de sodium (par le canal ENaC) et, dans le même temps, la sécrétion dans les urines de potassium (par le canal ROMK). De manière physiologique, l’hyperkaliémie stimule la sécrétion d’aldostérone et, à l’inverse, l’hypokaliémie peut la freiner. La sécrétion d’aldostérone entraîne une excrétion potassique urinaire réduisant la kaliémie, et son blocage diminue l’excrétion potassique urinaire, augmentant la kaliémie. Les principales causes d’anomalie de sécrétion de l’aldostérone sont les suivantes :
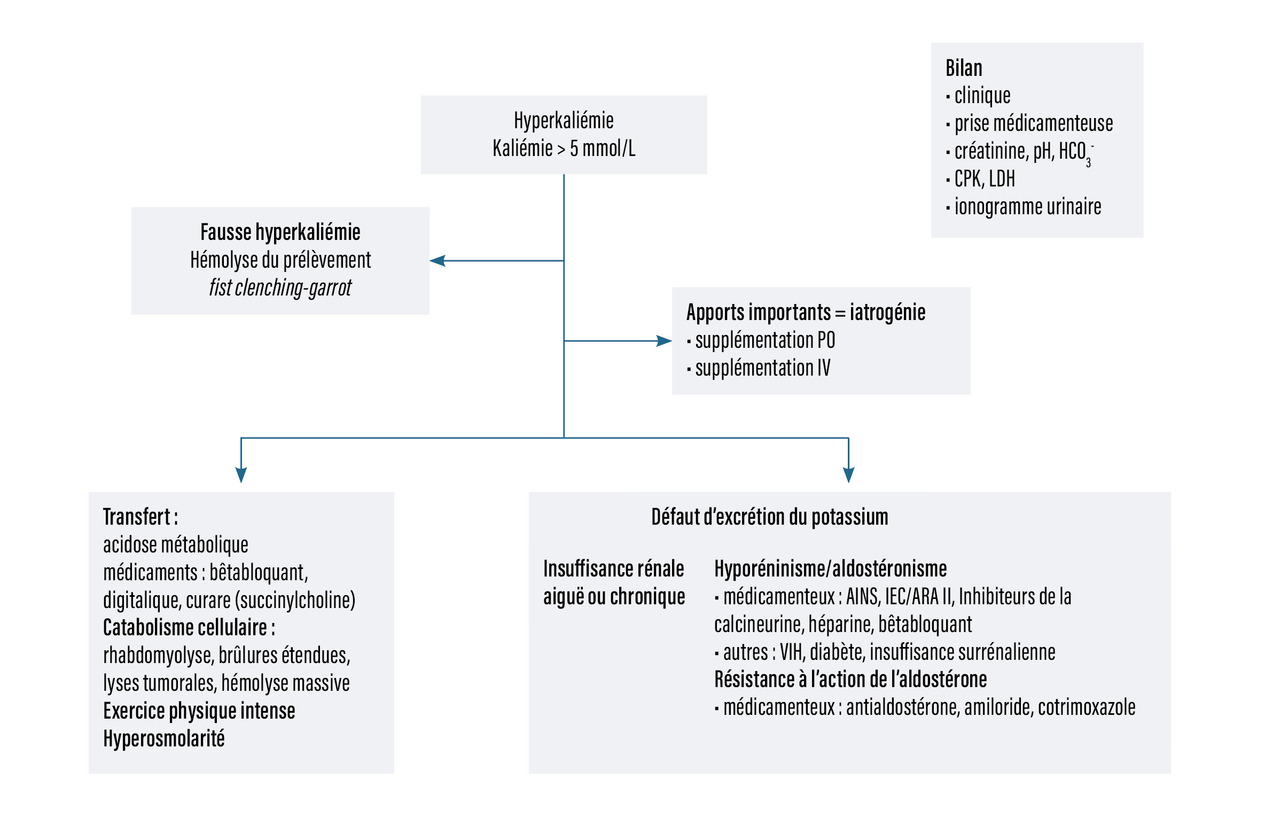
- hypoaldostéronisme par insuffisance surrénalienne, syndrome d’hyporéninisme hypo-aldostéronisme (VIH, diabète), prise d’anti-inflammatoires non stéroïdiens (via un hyporéninisme), d’inhibiteurs de la calcineurine (tacrolimus, ciclosporine), d’héparine, d’inhibiteurs de l’enzyme de conversion (IEC)/antagonistes des récepteurs de l’angiotensine II (ARAII) ;
- résistance à l’action de l’aldostérone par la prise d’antagonistes du récepteur de l’aldostérone (spironolactone, éplérénone), d’inhibiteurs du canal ENaC (amiloride), triméthoprime, pentamidine) ;
- hyperaldostéronisme, qui peut être primitif ou secondaire (v. infra).
Le récepteur à l’aldostérone n’est pas spécifique des minéralocorticoïdes, il est aussi sensible aux glucocorticoïdes. L’activation du récepteur par les glucocorticoïdes est empêchée par l’action d’une enzyme, la 11β -hydroxydéshydrogénase de type 2 (11βHSD de type 2). Cette enzyme métabolise le cortisol en cortisone inactive et empêche ainsi une surstimulation du récepteur. L’activité de cette enzyme peut être dépassée en cas de prise de corticoïdes exogène ou de syndrome de Cushing ; inhibée (par la glycyrrhizine contenue dans la réglisse ou le pastis sans alcool) ou être constitutionnellement diminuée (rares maladies génétiques). Lorsque l’activité de cette enzyme est diminuée ou dépassée, l’activation du récepteur de l’aldostérone est favorisée, conduisant à un tableau de pseudo-hyperaldostéronisme.
Les cellules : le potassium étant très majoritairement contenu en intracellulaire, toute augmentation importante du catabolisme cellulaire peut entraîner une hyperkaliémie : rhabdomyolyse, écrasement musculaire, hémolyse massive, lyse tumorale, cytolyse hépatique majeure, etc. À l’opposé, une prolifération cellulaire intense peut occasionner une hypokaliémie : stimulation de l’hématopoïèse par des facteurs de croissance, correction d’une carence en vitamine B9 ou B12, leucémie d’évolution rapide, etc.
De fausses dyskaliémies peuvent exister. Plusieurs facteurs peuvent artificiellement faire augmenter la kaliémie (par transfert du secteur intracellulaire vers le secteur extracellulaire) : un prélèvement hémolysé (car fait avec garrot, ou analysé trop longtemps après sa réalisation), la contraction musculaire réflexe des muscles de l’avant-bras lors du prélèvement (fist clenching). De fausses hypokaliémies sont décrites en cas d’hyperleucocytose majeure dans un contexte de pathologie hématologique.
Hyperkaliémie (rang A)
La démarche diagnostique étiologique de l’hyperkaliémie est résumée dans la fig. 3.
Retentissement clinico-biologique
Le retentissement de l’hyperkaliémie est principalement objectivable sur l’électrocardiogramme (ECG). Les situations menant à une hyperkaliémie symptomatique sur le plan clinique sont graves et souvent liées à des hyperkaliémies très sévères. En plus de la concentration plasmatique, la rapidité d’installation de l’hyperkaliémie favorise sa gravité. Les patients habitués à présenter des kaliémies hautes (typiquement les patients dialysés chroniques en interséance) tolèrent mieux l’hyperkaliémie.
Cliniquement, on peut observer des signes neuromusculaires, avec des anomalies de la sensibilité, des paresthésies des extrémités, une atteinte motrice avec une faiblesse des extrémités, voire une défaillance respiratoire et une paralysie flasque (signe de gravité extrême). Dans les formes sévères, l’atteinte cardiaque peut mener à une hypotension artérielle, constituant un signe de gravité.
L’hyperkaliémie entraîne une hypoexcitabilité myocardique avec un retentissement à l’ECG :
- signe précoce d’hyperkaliémie avec ondes T amples, symétriques et diffuses ;
- signes de gravité :
- allongement de l’espace PR puis disparition de l’onde P ;
- élargissement des QRS ;
- arythmie ventriculaire en cas d’association à d’autres troubles ioniques (hypocalcémie), d’acidose profonde, d’ischémie myocardique ;
- apparition d’un aspect sinusoïdal (sine wave pattern).
En l’absence de prise en charge, cette hypoexcitabilité myocardique évolue typiquement vers l’arrêt cardiaque par passage d’une bradycardie à QRS larges à une asystolie.
Traitement symptomatique général
En cas de signes de gravité à l’ECG, de signes neuromusculaires ou si la kaliémie est très élevée (supérieure à 7 mmol/L), la prise en charge constitue une urgence thérapeutique :
- gluconate de calcium 1 g, 1 à 2 ampoules en intraveineuse lente (IVL, 5 ou 10 min) ; son administration permet un effet stabilisateur de membrane et la correction des troubles de conduction mais ne fait pas baisser la kaliémie ;
- en cas d’intoxication digitalique, les sels de calcium sont contre-indiqués (utiliser le chlorure de magnésium) ;
- insuline-glucose avec 10 à 15 UI d’insuline rapide diluées dans 500 mL de glucose 10 %, à administrer en trente à quarante-cinq minutes par voie intraveineuse ; permet d’abaisser la kaliémie ;
- aérosol de salbutamol avec 20 mg de salbutamol (4 ampoules de 5 mg) en nébulisation, en même temps que l’insuline-glucose (effet synergique par stimulation de la Na-K-ATPase cellulaire, qui permet l’entrée de K+ dans la cellule en échange de Na+) ;
- bicarbonate de sodium en cas d’acidose métabolique, et uniquement en l’absence de surcharge volémique (risque d’œdème aigu pulmonaire [OAP] par l’apport en sodium). L’effet obtenu en cas d’hyperkaliémie menaçante est principalement un effet stabilisateur de membrane lié aux apports massifs en sodium ;
- diurétiques de l’anse qui permettent l’augmentation de l’excrétion rénale de potassium en cas de surcharge hydrosodée associée à l’hyperkaliémie (efficacité faible et lente) ;
- épuration extrarénale par hémodialyse à envisager systématiquement en association aux mesures précédentes en cas d’hyperkaliémie grave associée à une IRA ou IRC avancée.
En dehors du contexte de l’urgence (par exemple en cas d’hyperkaliémie modérée en ville, encadré 2), une résine échangeuse d’ions est utilisée par voie orale ou rectale. Son effet est faible et lent et elle est fréquemment utilisée chez les patients avec une IRC avancée ou en dialyse. Ce n’est pas un traitement de l’urgence ni de l’hyperkaliémie menaçante.
Traitements spécifiques
La première étape consiste en l’arrêt des traitements favorisant l’hyperkaliémie (IEC/ARAII [qui peuvent être diminués ou suspendus lors d’une hyperkaliémie mais dont le bénéfice global est souvent en faveur de leur maintien au long cours], inhibiteurs de calcineurine, cotrimoxazole, anti-inflammatoires non stéroïdiens, etc.).
En hospitalisation, il faut s’assurer de l’absence d’apport iatrogène de potassium. Dans la grande majorité des cas, ces mesures suffisent au contrôle de la kaliémie.
En cas de maladie rénale (aiguë ou chronique), du fait de la réduction de l’excrétion urinaire, les apports alimentaires peuvent jouer un rôle important. Une prise en charge diététique en situation d’hyperkaliémie modérée doit être proposée. En cas d’IRA/IRC sévère, selon le chiffre de la kaliémie, la persistance ou non d’une diurèse et la cause de l’insuffisance rénale, il faut discuter rapidement avec le spécialiste (néphrologue ou réanimateur) de l’indication à une épuration extrarénale en urgence.
En cas d’intoxication aux digitaliques, un traitement par anticorps spécifiques doit être administré (immunoglobulines ovines antidigitaliques).
Lors de l’insuffisance surrénalienne, l’hyperkaliémie est facilement contrôlée par la supplémentation en fludrocortisone.
Hypokaliémie
Bilan étiologique
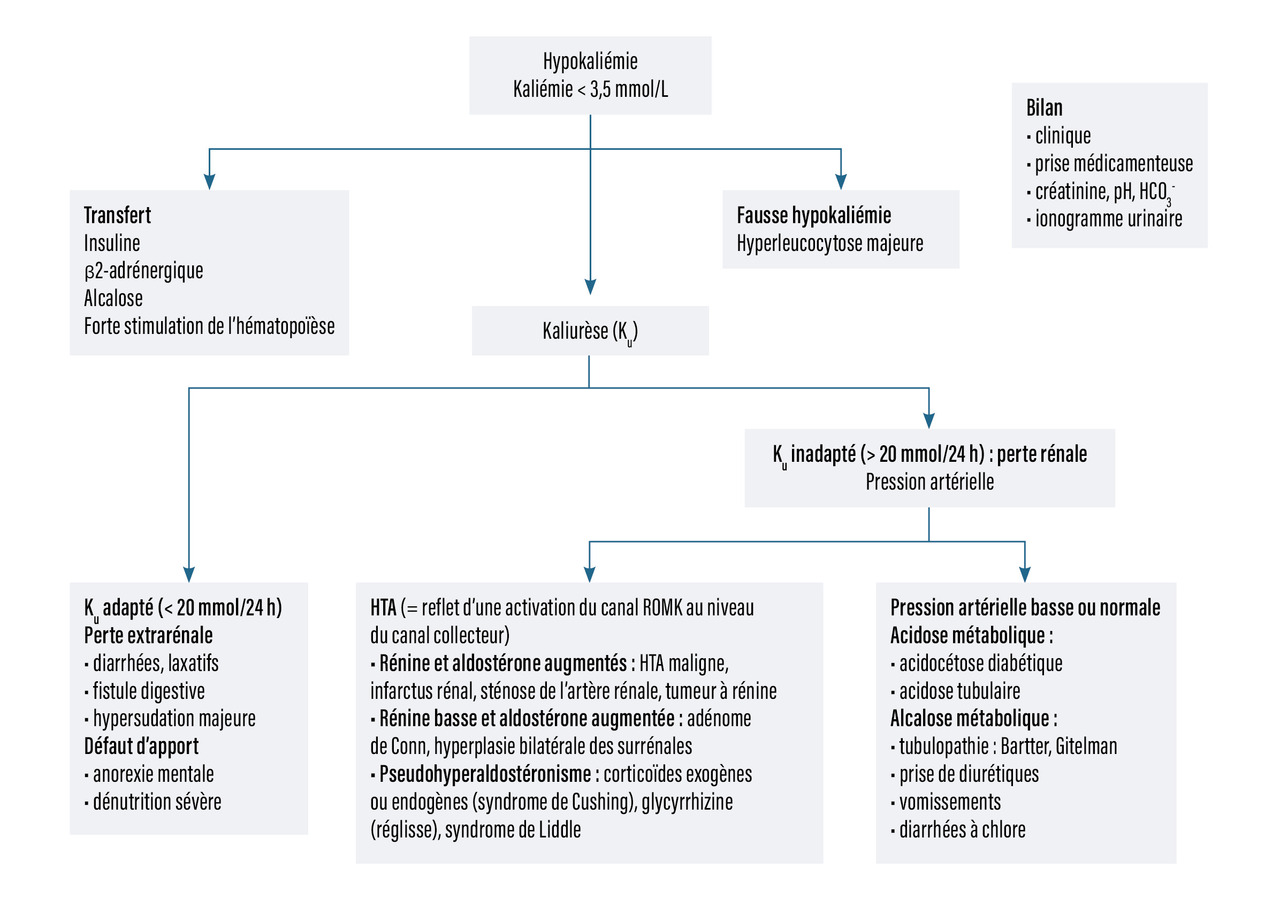
La démarche diagnostique de l’hypokaliémie est résumée dans la figure 4.
Quelques causes méritent d’être précisées.
Les hypokaliémies accompagnées d’une hypertension artérielle (HTA) doivent faire rechercher une cause secondaire d’HTA et peuvent être le reflet d’une hyperactivation du canal ROMK. Cette hyperactivation peut être liée soit à une sécrétion d’aldostérone (primitive ou secondaire), soit à une activation indépendante de l’aldostérone (activation constitutionnelle d’ENaC, du récepteur de l’aldostérone…) [fig. 5].
L’hypokaliémie concomitante d’une acidocétose diabétique : dans l’acidocétose diabétique inaugurale, le patient présente de manière systématique une carence en potassium intracellulaire (hypokalicytie). Cette carence provient de la fuite urinaire en potassium secondaire à la polyurie osmotique. Cette hypokalicystie est souvent masquée au début de la prise en charge par l’acidose mais se révèle de manière très précoce avec la correction de la carence en insuline. Ceci explique la supplémentation potassique quasi systématique et souvent majeure nécessaire lors de la prise en charge initiale des acidocétoses.
L’hypertension artérielle liée à la prise de corticoïdes est ainsi secondaire à une stimulation du récepteur de l’aldostérone (avec un profil d’hyperaldostéronisme-hyporéninisme biologique) par les corticoïdes exogènes (dépassant la capacité de métabolisation de la 11βHSD). Cela explique à la fois l’hypertension artérielle, la surcharge hydrosodée et l’hypokaliémie (par activation d’ENaC et de ROMK) lors de la prise d’un traitement par corticoïdes.
Retentissement clinico-biologique
Comme pour l’hyperkaliémie, les symptômes cliniques de l’hypokaliémie sont rares et témoignent fréquemment d’une hypokaliémie sévère.
Cliniquement, l’hypokaliémie sévère peut entraîner des troubles neuromusculaires à type de crampes, myalgies, paralysie. Au niveau digestif, elle est responsable d’une constipation. De manière chronique, l’hypokaliémie peut se compliquer d’une néphropathie hypokaliémique de profil tubulo-interstitiel (le plus souvent chez des patients avec mésusage chronique de diurétiques comme dans l’anorexie mentale).
Selon la profondeur de l’hypokaliémie, l’ECG montre un aplatissement de l’onde T, un allongement du QT, l’apparition d’une onde U (suivant l’onde T) jusqu’à un sous-décalage du segment ST. Cette hyperexcitabilité cardiaque ne doit pas être négligée, une hypokaliémie profonde peut provoquer le décès tout comme une hyperkaliémie (et peut être même plus grave que l’hyperkaliémie chez les patients avec une cardiopathie sous-jacente). Les troubles du rythme supraventriculaire peuvent être responsables d’un accident vasculaire cérébral embolique ; une torsade de pointes ou un autre trouble du rythme ventriculaire grave peut entraîner un arrêt cardiaque. La découverte d’un trouble du rythme supraventriculaire doit toujours faire rechercher une hypokaliémie.
Traitement symptomatique général
En cas de retentissement clinique ou paraclinique (signes ECG), de kaliémie inférieure à 2,5 mmol/L (absence de consensus) ou en cas d’intolérance alimentaire, une recharge intraveineuse par chlorure de potassium est réalisée, avec une concentration maximale de 4 g/L sur voie périphérique, à un débit maximal de 1 g/h (à diluer dans du NaCl plutôt que du glucose pour éviter une sécrétion d’insuline réactionnelle, qui peut aggraver l’hypokaliémie). En unité de soins intensifs, une recharge sur cathéter central permet une supplémentation dans un volume beaucoup plus faible (3 g dans 50 cc en trois heures maximum) mais nécessite une administration scopée et régulée par un pousse-seringue électrique. Le monitorage cardiaque continu est indiqué si l’hypokaliémie est profonde du fait du risque de troubles du rythme cardiaques graves. Dans tous les cas, s’il n’y a pas d’intolérance digestive, la recharge intraveineuse doit être complétée par une recharge orale.
En cas d’hypokaliémie asymptomatique, la recharge se fait par voie orale avec du chlorure de potassium : la quantité de potassium prescrite dépend de la profondeur de l’hypokaliémie et de sa cause. Dans tous les cas, une surveillance ionique rapprochée doit être réalisée après supplémentation (après introduction, après changement de dose, à l’arrêt de la supplémentation).
Une recherche de la cause de l’hypokaliémie et son traitement doivent toujours être associés à ces mesures.
Traitements spécifiques (rang B)
Plusieurs causes d’hypokaliémie nécessitent une prise en charge particulière.
Hyperaldostéronisme primaire : en cas d’adénome sécrétant latéralisé objectivé, la chirurgie est souvent le traitement recommandé. En l’absence ou en l’attente d’une chirurgie, un traitement par spironolactone est particulièrement efficace.
Le syndrome de Liddle est une maladie génétique rare caractérisée par une hypertension artérielle et une hypokaliémie très précoce secondaire à une activation constitutionnelle de ROMK au niveau du canal collecteur. Cette pathologie est importante à reconnaître car le traitement par amiloride permet une normalisation de la pression artérielle et de la kaliémie.
En cas de sténose d’une artère rénale, une intervention endovasculaire est rarement nécessaire, et cette indication est posée après avis en centre spécialisé. Si la tolérance le permet, un traitement par IEC/ARAII peut être efficace.
Dyscalcémie
Quelques éléments de physiologie
Le calcium est principalement stocké dans l’os et sa concentration plasmatique est finement régulée pour éviter les effets nocifs d’une dyscalcémie. Seule l’hypocalcémie est abordée dans cet item.
Les acteurs clés de la régulation de la calcémie sont les suivants :
- l’hormone parathyroïdienne (PTH), qui est synthétisée par les parathyroïdes en réponse à une hypocalcémie. Elle permet une augmentation de la calcémie par augmentation de la réabsorption du calcium au niveau rénal (tube contourné distal) et augmentation de la résorption osseuse. Elle stimule la synthèse du calcitriol et inhibe la réabsorption du phosphate au niveau rénal ;
- les récepteurs du calcium ou CaSR (calcium sensor), qui sont situés sur les cellules parathyroïdiennes, dans l’os et au niveau du tubule rénal. L’activation du CaSR au niveau de la branche large ascendante de Henlé (BLAH) entraîne une inactivation de la pompe Na-K- 2Cl, conduisant à une hypercalciurie, une natriurèse et donc à une hyperdiurèse. L’activation du CaSR sur les cellules parathyroïdiennes lors d’une hypercalcémie bloque la sécrétion de PTH ; au contraire, l’hypocalcémie stimule la sécrétion de PTH ;
- le calcitriol (ou 1 - 25 OH vitamine D3 ou vitamine D active), qui augmente l’absorption digestive calcique, bloque la PTH et stimule le FGF23. Il augmente également l’absorption digestive du phosphate. Il est issu de l’hydroxylation de la 25 -OH vitamine D2 par la 1 -alphahydroxylase exprimée par le tubule proximal rénal ;
- la calcémie totale, qui correspond au calcium ionisé additionné au calcium fixé aux protéines (très majoritairement l’albumine). Cette fixation dépend du pH. C’est la calcémie ionisée qui entraîne des effets biologiques. En cas d’hyper- ou d’hypoprotidémie, la calcémie totale n’est donc pas interprétable. Les formules de correction de la calcémie en fonction de la protidémie ne sont pas parfaitement rigoureuses. Il est toujours préférable de mesurer et d’interpréter la calcémie ionisée.
Hypocalcémie
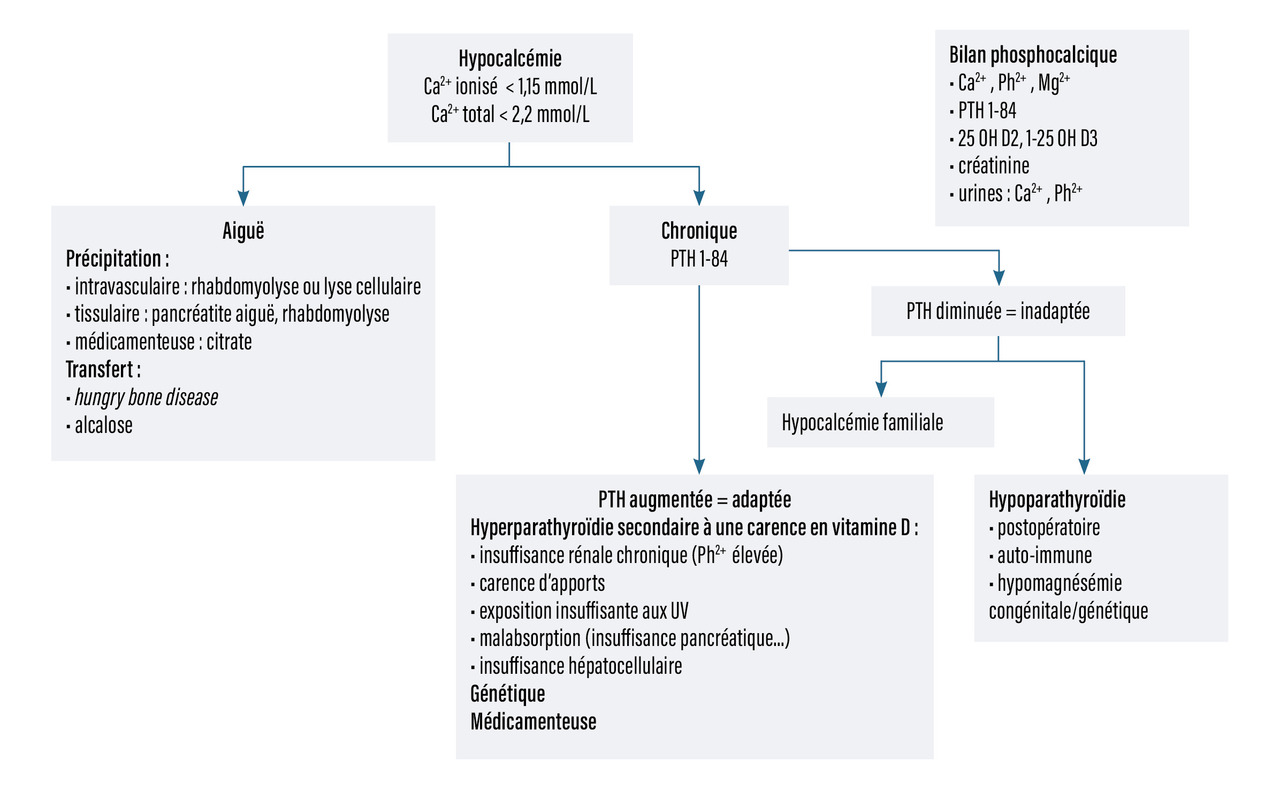
L’hypocalcémie (calcémie totale inférieure à 2,20 mmol/L ou calcémie ionisée inférieure à 1,15 mmol/L) est une anomalie métabolique peu fréquente qui résulte soit d’une augmentation des pertes de calcium (dépôt tissulaire, transfert vers l’os, perte urinaire ou chélation intravasculaire), soit d’une diminution des entrées de calcium dans la circulation (réduction de la résorption osseuse ou malabsorption intestinale).
Bilan étiologique
La démarche diagnostique étiologique de l’hypocalcémie est résumée dans la figure 6.
Les trois causes les plus fréquentes en sont l’hypoparathyroïdie, la carence en vitamine D et l’insuffisance rénale chronique.
Hypoparathyroïdie
Il s’agit d’une diminution primitive de la PTH, entraînant une hypocalcémie, qui peut avoir des origines multiples :
- postopératoires : la plus fréquente, des suites d’une chirurgie ORL ;
- infiltration parathyroïdienne : cancer, amylose, granulome, lymphome ;
- l’hypomagnésémie entraîne une diminution de la production de PTH mais aussi une résistance périphérique à son action (pseudo-hypoparathyroïdie) ;
- des maladies génétiques peuvent être responsables d’hypocalcémie, avec profil de PTH adaptée ou inadaptée (syndrome de DiGeorge). Ces causes s’intègrent souvent dans le cadre de pathologies avec atteintes d’organes multiples.
Carence en vitamine D
Elle peut avoir plusieurs origines :
- carences d’apports (essentiellement chez les enfants) ;
- exposition insuffisante aux ultraviolets ;
- malabsorption des graisses responsable d’une malabsorption des vitamines liposolubles (dont la vitamine D) ;
- insuffisance rénale chronique (encadré 3).
Hypocalcémie hypercalciurique familiale
Cette entité est secondaire à une mutation activatrice du CaSR. Le CaSR est continuellement activé, responsable d’une PTH basse, d’une hypercalciurie et d’une hypocalcémie en général modérée.
Hungry bone disease
Lors d’une parathyroïdectomie pour hyperparathyroïdie primaire ou tertiaire prolongée, la correction brutale de la PTH (qui devient alors basse ou normale) peut entraîner une accrétion calcique osseuse brutale, cause d’une hypocalcémie aiguë. Cette hypocalcémie peut être très profonde et nécessiter une supplémentation intraveineuse et per os, associée à une supplémentation en vitamine D native et active. En pratique, la guérison de toute ostéopathie métabolique intense peut entraîner ce syndrome.
Causes médicamenteuses
Les mécanismes principaux sont une interaction avec le métabolisme de la vitamine D, ou action directe sur des régulateurs du métabolisme calcique. Les médicaments le plus fréquemment à l’origine d’une hypocalcémie sont les corticoïdes, certains anticonvulsivants (augmentation de dégradation de la vitamine D par le cytochrome CYP24A1), certaines antiprotéases (diminution de la 25 -hydroxylation hépatique), le citrate (chélation du calcium) utilisé pour l’anticoagulation locale d’un circuit extracorporel, les calcimimétiques (mal employés ou surdosés), les bisphosphonates (qui favorisent la fixation du calcium à l’os).
Alcalose
Elle entraîne une augmentation de la fixation du calcium libre aux protéines, ce qui diminue la calcémie ionisée. Lors d’une attaque de panique, l’alcalose respiratoire aiguë secondaire à l’hyperventilation est responsable d’une hypocalcémie « efficace », responsable de la tétanie.
Situations particulières
Dans certaines situations aiguës, le calcium peut précipiter dans les tissus (rhabdomyolyse, pancréatite aiguë) ou en intravasculaire (lyse tumorale, rhabdomyolyse) et entraîner une hypocalcémie aiguë.
Retentissement
Cliniquement, on retrouve des manifestations neuromusculaires : paresthésies souvent au premier plan (pourtour des lèvres, doigts), spasmes (parfois jusqu’à la crise de tétanie), troubles des fonctions supérieures, voire des convulsions. Deux signes cliniques sont classiquement décrits :
- le signe de Chvostek, qui correspond à une contraction faciale lors de la percussion du nerf facial ;
- le signe de Trousseau, qui est une contraction de la main, dite en « main d’accoucheur », lors de la compression de l’artère brachiale (par un brassard à tension, par exemple).
L’hypocalcémie entraîne, à l’ECG, un allongement du QT, pouvant favoriser certains troubles du rythme.
Elle peut s’associer dans certains cas à une hypomagnésémie, qu’il faut penser à rechercher.
Traitement symptomatique
En cas de retentissement clinique ou paraclinique : recharge intraveineuse (gluconate de calcium ou chlorure de calcium) à associer à la supplémentation orale (carbonate de calcium). La supplémentation en phosphate ne doit pas être réalisée dans la même perfusion intraveineuse du fait du risque de précipitation.
En cas de forme asymptomatique :
- augmentation de la prise alimentaire (produits laitiers, eaux riches en calcium ;
- recharge médicamenteuse par calcium élément, à prendre en dehors des repas (favorise l’absorption car sinon le calcium se complexe au phosphate alimentaire et est éliminé dans les selles), associé à la correction d’une carence en vitamine D.
La supplémentation en vitamine D active est rarement nécessaire et est en général réservée aux insuffisances rénales chroniques avancées et lors d’hypoparathyroïdies sévères.
Il convient de corriger en parallèle l’hypomagnésémie, si elle est présente.
Dans le cas particulier de la rhabdomyolyse, l’hypocalcémie doit plutôt être respectée (sauf si elle est symptomatique), car sa correction favorise la précipitation calcique.
Troubles acidobasiques
Il existe deux sources d’acide dans l’organisme : les acides volatils provenant du métabolisme cellulaire (CO2), et les acides fixes (H+) issus du métabolisme des protéines (donc de l’alimentation).
Le pH reflète la concentration d’ions H+ dans une solution. Le pH physiologique est toujours compris entre 7,38 et 7,42. La pression partielle de dioxyde de carbone (PaCO2) est comprise entre 35 et 45 mmHg. La valeur normale des bicarbonates (HCO3 -) est comprise entre 22 et 26 mmol/L.
La valeur du pH est très finement régulée par trois acteurs :
- le système de tampon bicarbonate, qui permet par exemple de tamponner immédiatement les acides apportés par l’alimentation selon la formule : H+ + HCO3- = H2CO3 = CO2 + H2O ;
- le système respiratoire, qui permet, par la ventilation alvéolaire, d’éliminer l’acide. La PaCO2 reflète la ventilation alvéolaire : lorsque la PaCO2 est supérieure à 45 mmHg, il existe une hypoventilation alvéolaire ; lorsque la PaCO2 est inférieure à 35 mmHg, il existe une hyperventilation alvéolaire ;
- le rein, qui permet de régénérer du bicarbonate (ce qui équivaut à éliminer un H+ au niveau du tube contourné distal) et réabsorber le bicarbonate filtré (au niveau du canal collecteur).
Il est d’usage de parler du pH artériel, mais le pH veineux est aussi utilisable, avec des normes différentes (pH physiologique compris entre 7,32 et 7,38).
L’acidose métabolique correspond à une baisse des bicarbonates, l’acidose respiratoire à une augmentation de la PaCO2, l’alcalose métabolique à une augmentation des bicarbonates, l’alcalose respiratoire à une baisse de la PaCO2.
Physiologiquement, en cas d’apparition d’un trouble acidobasique primitif, l’organisme tente toujours de le corriger dans la limite de ses capacités (tableau). La compensation respiratoire d’un trouble métabolique est immédiate, tandis que la compensation rénale métabolique d’un trouble acidobasique respiratoire peut prendre vingt-quatre à quarante-huit heures.
L’apparition d’un trouble décompensé (pH inférieur à 7,38 ou supérieur à 7,42) témoigne d’un dépassement des capacités d’adaptation de l’organisme. L’acidémie correspond à une acidose décompensée entraînant une baisse du pH (inférieur à 7,38). L’alcalémie correspond à une alcalose décompensée entraînant une augmentation du pH (supérieur à 7,42).
Le pH (extracellulaire), les PaO2 et PaCO2 (à partir desquelles le HCO3- est calculé) se mesurent à l’aide d’une gazométrie qui peut être réalisée en cas de dyspnée ou de suspicion de pathologie hypoxémiante, de suspicion clinique de trouble acidobasique, d’état de choc, d’insuffisance rénale aiguë sévère. Cette gazométrie se réalise par un prélèvement sanguin sur une seringue à gaz du sang, de préférence artériel, classiquement au niveau radial.
Il est possible d’avoir un trouble acide-base dit simple (acidose/alcalose uniquement métabolique ou respiratoire) ou mixte, c’est-à-dire une participation métabolique et respiratoire. Un trouble complexe correspond, par exemple, à l’association d’une acidose métabolique à une alcalose respiratoire.
Acidose métabolique primitive
Bilan étiologique
Le trouble primitif est métabolique (baisse de la bicarbonatémie, HCO3- inférieur à 22 mmol/L) et peut être compensé par une augmentation de l’excrétion respiratoire des acides (c’est-à-dire une baisse de la PaCO2). La PaCO2 attendue est égale à 1,5 × [HCO3-] + 8 (± 2). Si la PaCO2 est plus élevée qu’attendue, il faut suspecter un trouble mixte ; si elle est plus basse, un trouble complexe.
Devant une acidose métabolique, l’urgence diagnostique est au calcul du trou anionique (TA) plasmatique. Le TA correspond à la différence entre les principaux cations et anions plasmatiques (la différence de tous les anions et cations, non réalisée en pratique, serait nulle selon le principe d’électroneutralité du plasma). Selon la prise en compte de la kaliémie, le TA normal varie entre 12 ± 4 (sans la kaliémie) et 16 ± 4 (avec la kaliémie).
TA = Na+ + K+ – ( Cl- + HCO3 -) = 16 ± 4
Un TA augmenté suggère la présence d’un ou plusieurs anions indosés associés à l’ion H+. Cela oriente toujours vers une cause pouvant menacer le pronostic vital : insuffisance rénale (plusieurs anions indosés comme les acides organiques, sulfates, etc.), acidocétose diabétique (les anions sont les corps cétoniques apparaissant lors d’une carence en insuline), acidose lactique (le lactate lié le plus souvent à une hypoxie cellulaire, une intoxication à la metformine...), certaines intoxications par un acide exogène (salicylate, éthylène glycol, méthanol) et acidocétose euglycémique liée aux inhibiteurs du SGLT2 (encadré 4).
Un TA normal ou bas suggère une perte de tampon bicarbonate par gain d’acide chlorhydrique (HCl), soit d’origine digestive (diarrhées ; la réponse rénale est alors normale), soit d’origine rénale (groupe des acidoses tubulaires).
Retentissement clinicobiologique
L’urgence est représentée par la profondeur de l’acidose et sa cause : en cas de pH inférieur à 7,10 ou de bicarbonatémie inférieure à 8 mmol/L, il existe une menace vitale. La symptomatologie est variable : diminution des débits cardiaques et tissulaires, résistance aux catécholamines, arythmies ventriculaires, inhibition du métabolisme cellulaire, coma, etc.
Traitement (rang B)
Le traitement d’une acidose métabolique est principalement celui de la cause : insulinothérapie pour l’acidocétose diabétique, épuration extrarénale pour l’insuffisance rénale organique sévère, traitement de l’état de choc, supplémentation en bicarbonates (uniquement indiquée dans les acidoses hyperchlorémiques par perte de bicarbonates).
Acidose respiratoire primitive
L’acidose respiratoire est secondaire à une diminution de la capacité d’excrétion des charges acides par l’appareil respiratoire, ce qui correspond à une hypoventilation et donc une augmentation de la PaCO2 (hypercapnie). Les causes sont variées :
- hypoventilation d’origine pulmonaire par décompensation de bronchopneumopathie chronique obstructive (BPCO), réduction parenchymateuse (emphysème sévère, résection chirurgicale étendue, etc.), certaines pathologies inflammatoires ;
- atteinte de la pompe ventilatoire par altération de la fonction neuromusculaire : myasthénie, myopathie, sclérose latérale amyotrophique (SLA), syndrome de Guillain-Barré, botulisme, poliomyélite ;
- atteinte de la cage thoracique : cyphoscoliose, épanchement pleural, obésité ;
- atteinte du contrôle ventilatoire par dysfonction cérébrale : encéphalites, accident vasculaire cérébral du tronc, traumatismes,
- dysfonction des centres respiratoires : syndrome d’Ondine, hypothyroïdie, syndrome d’apnées du sommeil central, sédatifs, tétanos, lésion des voies afférentes et efférentes (sclérose en plaques, maladie de Parkinson, myélite, traumatisme cervical au-dessus de C5) ;
- dysfonction des récepteurs périphériques : syringomyélie, dysautonomie familiale, neuropathie diabétique, tétanos.
Les signes cliniques de l’hypercapnie sont les sueurs, les céphalées, une polypnée réactionnelle, des troubles de la vigilance pouvant aller jusqu’au coma.
Le traitement est celui de la cause (traitement de la décompensation de la BPCO, antibiothérapie, etc.) et la mise en place d’un support ventilatoire : ventilation invasive ou non invasive qui permet l’augmentation de la ventilation alvéolaire et donc l’élimination du CO2.
Alcalose métabolique primitive
Le trouble primitif est métabolique (augmentation de la bicarbonatémie, HCO3- supérieur à 26 mmol/L) et peut être compensée par une baisse de l’excrétion respiratoire des charges acides, soit une hypoventilation (augmentation de la PaCO2).
Ce trouble est principalement rencontré lors d’une alcalose de contraction induite par une déplétion trop importante par un diurétique.
Les facteurs favorisants sont l’hypovolémie réelle ou efficace (par activation secondaire du système rénine-angiotensine-aldostérone), une perte d’H+ digestive (vomissements, aspiration nasogastrique, diarrhées chlorées), un hyperminéralocorticisme.
Les facteurs d’entretien sont la persistance de l’hypovolémie, l’hypokaliémie, l’hyperminéralocorticisme, l’hypochlorémie.
Le traitement repose sur celui de la cause (correction volémique notamment) et les supplémentations ioniques.
Alcalose respiratoire primitive
Elle est liée à une hyperventilation qui se traduit par une inspiration profonde et/ou rapide. L’archétype est la dyspnée de Kussmaul (lors de la tentative de compensation d’une acidose métabolique profonde, typiquement dans les acidocétoses diabétiques).
Toute hyperventilation peut entraîner une alcalose respiratoire. Un exemple classique en est la crise d’asthme aiguë : typiquement, la PaCO2 est initialement basse, avec une alcalose possible. La normalisation de la PaCO2, voire son augmentation et l’apparition d’une acidose respiratoire, est un signe de gravité, témoignant de l’épuisement respiratoire du patient. L’hyperventilation peut aussi être liée à une crise d’angoisse (« spasmophilie »), la tétanie est secondaire à l’alcalose aiguë, entraînant une baisse de la calcémie ionisée (par fixation du calcium aux protéines), à une encéphalopathie (hépatique, par exemple) ou à une intoxication (notamment aux salicylés).
Dysnatrémie
La natrémie est le reflet de l’hydratation intracellulaire (bilan hydrique) ; la volémie (volume vasculaire) est le reflet de la quantité de sodium (bilan sodé).
L’hyponatrémie (Na+ < 135 mmol/L) correspond à une hyperhydratation intracellulaire (attention aux fausses hyponatrémies) ; l’hypernatrémie (> 145 mmol/L) à une déshydratation intracellulaire.
L’hypovolémie entraîne une réabsorption de Na+ (activation du SRAA) et une réabsorption d’eau (sécrétion d’ADH).
L’hypovolémie peut être « vraie » (déshydratation extracellulaire globale) ou « efficace » (hypovolémie artérielle mais possible hypervolémie veineuse et/ou interstitielle).
Dyskaliémie
L’hyperkaliémie provoque une hypo-excitabilité myocardique, soit un « étirement de l’ECG ». En ville, il faut penser aux fausses hyperkaliémies (par hémolyse d’un prélèvement ayant attendu longtemps), surtout quand elles sont modérées. Le traitement est à connaître parfaitement : gluconate de calcium en urgence pour le retentissement cardiaque et traitement hypokaliémiant par insuline-glucose, aérosols de salbutamol, arrêt des traitements hyperkaliémiants et, enfin, traitement de la cause. L’indication à une épuration extrarénale doit être reconnue : typiquement, hyperkaliémie avec insuffisance rénale oligo-anurique.
L’hypokaliémie entraîne une hyperexcitabilité myocardique et des troubles du rythme (supra)ventriculaire. La démarche diagnostique de l’hypokaliémie, notamment associée à une HTA, doit être connue ; le bilan étiologique différencie l’hyperaldostéronisme primaire (adénome de Conn et hyperplasie bilatérale des surrénales), l’hyperaldostéronisme secondaire (sténose des artères rénales, infarctus rénal, HTA maligne, tumeur à rénine) et le pseudo-hyperaldostéronisme (hypercorticisme endogène ou exogène, réglisse, syndrome de Liddle).
Hypocalcémie
Les signes cliniques classiques sont ceux de Trousseau et de Chvostek. Il existe également des signes non spécifiques (paresthésies). Un retentissement électrocardiographique est possible (troubles du rythme).
Les causes principales avec PTH basse (hypoparathyroïdie) et PTH haute (insuffisance rénale chronique, carence en vitamine D, pseudo-hypoparathyroïdie, hypocalcémie de transfert) doivent être différenciées.
Troubles acidobasiques
La réalisation d’un gaz du sang artériel est indispensable devant une suspicion de trouble acidobasique.
Il s’agit de savoir calculer le trou anionique et évoquer les causes d’acidose à trou anionique augmenté avec leur traitement de base. De même, il faut savoir reconnaître une acidose liée à une perte digestive.
Devant une alcalose métabolique, l’alcalose de contraction liée à un diurétique doit être évoquée.
1. Correction trop rapide d’une dysnatrémie
Qu’il s’agisse d’une hyponatrémie d’ancienneté indéterminée ou d’une hyponatrémie installée depuis plus de quarante-huit heures, la correction du trouble hydrique ne doit pas être trop rapide. En effet, les cellules neuronales ne sont pas perméables aux osmoles circulantes et le maintien de leur tonicité dépend d’un stock particulier, les osmoles osmogéniques. Lors d’une dysnatrémie, la cellule produit ou dégrade des osmoles osmogéniques (synthèse/dégradation en quarante-huit heures) pour maintenir une osmolarité cellulaire équivalente à celle du milieu extérieur. En cas d’hyponatrémie, la cellule perd son stock d’osmoles osmogéniques ; si la correction de la natrémie est trop rapide, il existera alors une hyperosmolarité extracellulaire et une hypo-osmolarité intracellulaire relative. Cette différence entraînera un mouvement d’eau du secteur intracellulaire vers l’extracellulaire responsable d’une myélinolyse centropontine. Il est parfois nécessaire de faire abaisser à nouveau l’osmolarité plasmatique si la correction initiale a été trop rapide pour l’éviter (par exemple avec de la vasopressine). À l’inverse, la littérature récente montre qu’il y a peu de risques d’une correction rapide de l’hyperosmolalité chez l’adulte (au contraire des enfants).
2. Hyperkaliémie modérée en ville (rang A)
L’hyperkaliémie modérée en médecine de ville est fréquente. En l’absence d’insuffisance rénale, cette hyperkaliémie est souvent fausse ou iatrogène. La première étape consiste à rechercher une hémolyse du prélèvement responsable d’une fausse hyperkaliémie (cf. introduction). Dans ces situations, un prélèvement de contrôle, réalisé au laboratoire, sans garrot est la première chose à faire. En pratique, une des causes fréquentes d’hyperkaliémie modérée est la prise d’IEC ou d’ARAII à visée antihypertensive ou cardio-/néphroprotectrice. Ces traitements sont souvent indispensables et leur arrêt peut augmenter la mortalité cardiovasculaire. Dans ces cas, l’ajout d’un diurétique (thiazidique ou de l’anse selon la situation) permet un meilleur contrôle de la kaliémie. Plus récemment, les inhibiteurs de sodium-glucose cotransporteur 2 (SGLT2), devenus un traitement majeur de l’insuffisance rénale chronique, du diabète et de l’insuffisance cardiaque, permettent souvent un meilleur contrôle de la kaliémie par leur action diurétique.
3. Hypocalcémie de l’insuffisance rénale chronique
Lors de l’insuffisance rénale chronique, le métabolisme phosphocalcique est fortement perturbé. La réduction néphronique entraîne une baisse de la 1 -hydroxylase, ce qui diminue la quantité de vitamine D active (calcitriol), réduisant l’absorption digestive de calcium. L’insuffisance rénale s’accompagne aussi d’une baisse de l’excrétion du phosphate qui est compensée initialement par une augmentation du FGF23, ayant comme effet secondaire une inhibition du calcitriol. L’hypocalcémie entraîne alors une stimulation de la PTH. Lorsque cette hyperparathyroïdie se prolonge (typiquement chez les patients avec insuffisance rénale chronique de stade V), un nodule parathyroïdien constitué de cellules sécrétant une grande quantité de PTH peut s’autonomiser (hyperparathyroïdie tertiaire), conduisant progressivement à une hypercalcémie associée à une PTH inadaptée. Le traitement consiste alors en la résection du nodule parathyroïdien ou en la prise de calcimimétiques.
4. Acidocétose euglycémique des gliflozines
Les gliflozines sont de plus en plus prescrites pour leur effet antidiabétique, néphroprotecteur et cardioprotecteur. L’acidocétose euglycémique est une complication rare mais grave de ces traitements. Elle se manifeste par une altération de l’état général associée biologiquement à une acidose métabolique pouvant être profonde et à une glycémie normale. La cétonémie est alors positive. Ces patients doivent être pris en charge comme s’ils étaient en acidocétose diabétique, en l’absence actuelle de prise en charge spécifique recommandée. La physiopathologie de cette acidose est encore mal connue, mais le nombre grandissant de patients traités par cette classe médicamenteuse oblige à connaître cette complication.
Drapeaux rouges
Troubles acidobasiques : ne pas rater l’acidose à trou anionique augmenté qui nécessite une prise en charge spécifique et induit un risque vital pour le patient.
Hyponatrémie : toujours s’assurer du caractère symptomatique ou non, ce qui modifie la prise en charge. S’assurer que l’hyponatrémie est hypotonique.
Hypernatrémie : l’eau libre et/ou le soluté glucosé constituent le traitement. La voie orale (eau libre) est utilisée, en association avec la voie intraveineuse (G5 %) si besoin.
Hyperkaliémie : l’ECG est indispensable et le traitement est une urgence en cas de signes cliniques/électrocardiographiques. La cause doit être recherchée et les traitements symptomatiques évités.
Hypokaliémie : toute hypokaliémie est à fort risque rythmique cardiaque, elle n’est jamais à négliger et sa cause doit toujours être recherchée ; les traitements substitutifs à l’aveugle sont à éviter.
Hypocalcémie : il est important de mesurer la calcémie en cas de trouble neurologique ou digestif et de ne pas raisonner sur la calcémie totale ou corrigée sur les protéines mais, si disponible, sur la calcémie ionisée.
Dans cet item très long, beaucoup de points peuvent être abordés lors de l’EDN. Nous détaillons ci-dessous certains des plus probables.
Troubles acidobasiques : le calcul du trou anionique plasmatique dans l’acidose métabolique et l’évocation, dans l’urgence, des trois causes principales d’acidose à trou anionique augmenté (acidocétose, acidose lactique et insuffisance rénale).
Hyponatrémie : la démarche diagnostique doit être parfaitement acquise. Le calcul de l’osmolalité plasmatique est la première étape et permet de s’assurer qu’il s’agit d’une « vraie » hyponatrémie (hypo-osmolaire). Il faut avoir bien compris les différences de traitement en cas d’hyponatrémie liée à une déshydratation extracellulaire globale (réhydratation), liée à une hypovolémie efficace (diurétiques) et liée à un SIADH (restriction hydrique et traitement de la cause). Le SIADH fait fréquemment l’objet de dossiers, notamment comme porte d’entrée dans la découverte d’une tumeur (pulmonaire le plus souvent). Toujours rechercher des signes de gravité, notamment neurologiques.
Hypernatrémie : le traitement est simple (réhydratation avec de l’eau), les questions s’intéresseront certainement davantage à des causes particulières, notamment le diabète insipide. Un dossier portant sur un diabète insipide secondaire à la prise de lithium est classique.
Hyperkaliémie : tout l’item doit être parfaitement connu car les questions sont fréquentes et nombreuses (reconnaissance d’un ECG typique d’hyperkaliémie, traitement des signes ECG [gluconate de calcium], traitement médicamenteux [en urgence] de l’hyperkaliémie).
Hypokaliémie : le point le plus probable est celui de la démarche diagnostique devant une HTA associée à une hypokaliémie. Il faut savoir, à partir d’un dosage de rénine et d’aldostérone, s’orienter vers les causes possibles de ce trouble.
Hypocalcémie : la reconnaissance des signes cliniques, notamment les signes de Trousseau et Chvostek, est essentielle. Les dossiers probables sont ceux d’une carence en vitamine D, de la découverte d’une maladie rénale chronique ou d’une hypoparathyroïdie. Il est important de bien raisonner sur la calcémie ionisée.
Sterns RH. Disorders of Plasma Sodium — Causes, Consequences, and Correction. Ingelfinger JR, ed. N Engl J Med. 2015;372(1):55-65. doi:10.1056/NEJMra1404489
Collège des enseignants d’endocrinologie, diabète et maladies métaboliques. https://www.sfendocrino.org/polycopie-des-enseignants-5eme-edition-2021/
Long B, Lentz S, Koyfman A, Gottlieb M, et al. Euglycemic diabetic ketoacidosis: Etiologies, evaluation, and management. Am J Emerg Med 2021;44:157-60.
 Encadrés
Encadrés