Les technologies de séquençage à haut débit (SHD) ont été mises au point à la fin des années 1990. Elles ont été développées dans des projets de recherche tels que le séquençage du génome humain1, puis la caractérisation des anomalies récurrentes des génomes tumoraux par des consortiums internationaux. Depuis les années 2010, de nouvelles générations d’automates sont devenues accessibles à l’ensemble des laboratoires, ce qui a permis leur implémentation dans la prise en charge diagnostique de routine dans de nombreux domaines et particulièrement en cancérologie.2 L’accumulation de mutations somatiques dans des gènes dits « drivers » confère aux cellules un avantage sélectif et participe, par un processus multi-étape, au développement des tumeurs. La caractérisation précise des événements génétiques ayant contribué au développement tumoral permet de retracer l’histoire évolutive du cancer et d’en déduire ses caractéristiques phénotypiques d’intérêt clinique. Ainsi, l’analyse des mutations somatiques occupe désormais un rôle majeur dans la prise en charge du patient pour le diagnostic, la nosologie, la détermination du pronostic, le choix thérapeutique et permet également de suivre l’évolution tumorale sous traitement, ouvrant la voie à des stratégies d’adaptation thérapeutique rationnelles. De plus, le SHD permet de mettre en évidence des variants germinaux de prédisposition qui peuvent conditionner le choix d’un donneur en cas d’allogreffe et mener à un conseil génétique chez les apparentés.
Le SHD « pour les nuls »
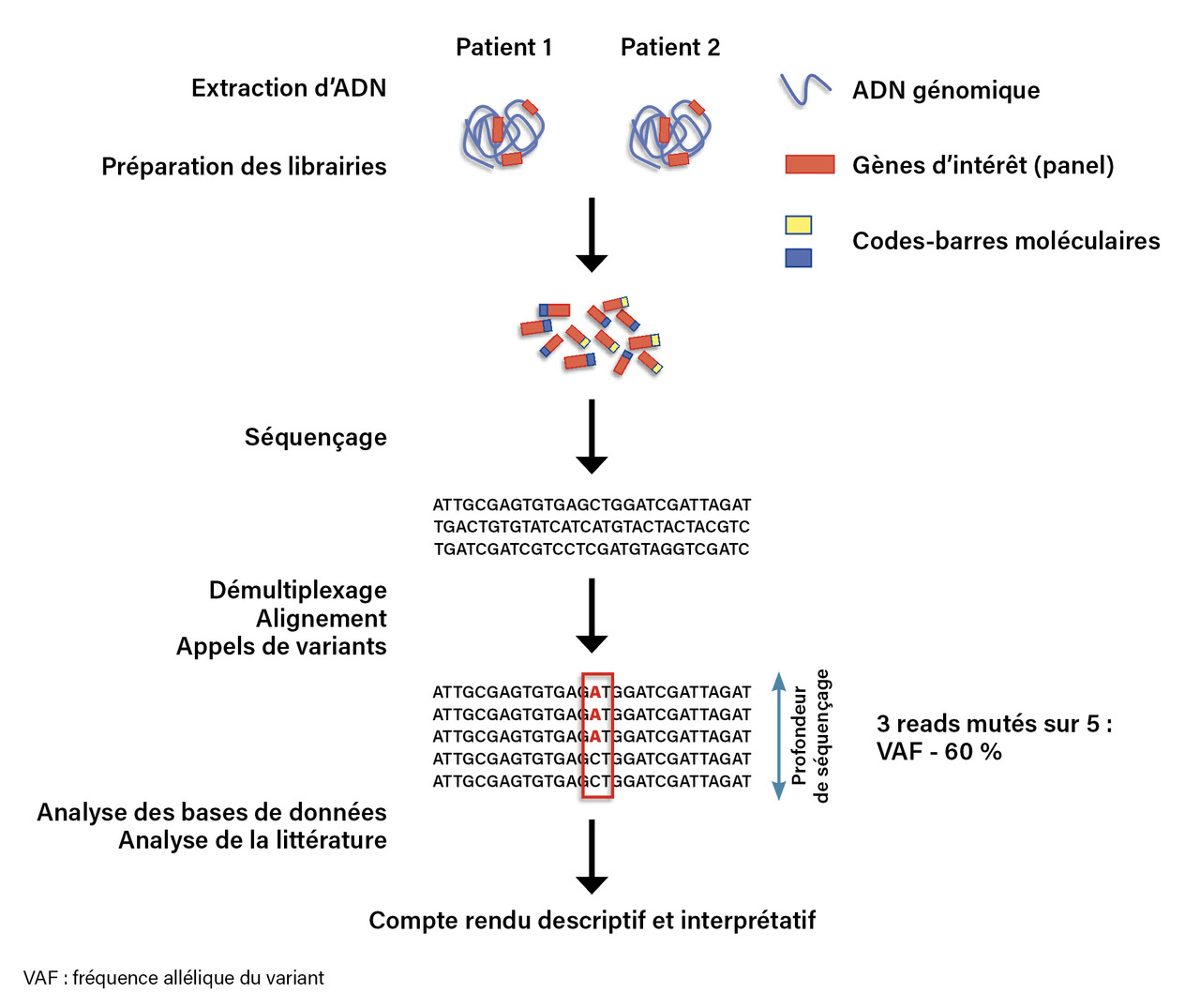
Comme pour tout acte de biologie médicale, la phase pré-analytique joue un rôle majeur dans la qualité du résultat final. En hématologie, le tissu de départ est généralement le sang ou la moelle osseuse, voire du tissu biopsique (ganglion, etc.) et, plus récemment, l’ADN libéré par les cellules tumorales dans le plasma (ADN tumoral circulant). Le degré d’envahissement par les cellules tumorales est un élément majeur : le seuil de détection d’une anomalie relevante par les techniques de SHD étant de l’ordre de 1 %, il serait illusoire de les utiliser dans des tissus trop peu envahis (lymphome de Hodgkin, par exemple). Les techniques d’extraction des acides nucléiques sont désormais largement standardisées et automatisées. Elles permettent d’obtenir une matrice de qualité satisfaisante à condition que des délais d’acheminement raisonnables puissent être respectés (figure).
Une fois l’ADN obtenu, l’étape de préparation des librairies (par amplification et/ou capture) permet de ne séquencer que les zones du génome qui ont un intérêt clinique : il s’agit du panel de gènes défini en amont par les biologistes.3 L’optimisation du panel est une étape clé : celui-ci doit être suffisamment exhaustif pour répondre à toutes les questions cliniques mais suffisamment petit pour que le coût de l’analyse reste raisonnable.4 Les librairies de chaque patient sont marquées par un code-barres moléculaire spécifique, puis elles sont mélangées (multiplexage) et déposées sur des puces, qui permettent le séquençage par synthèse d’un brin d’ADN complémentaire : une polymérase recopie la séquence, et émet un signal spécifique (lumineux ou électrique selon le fournisseur) à chaque nucléotide complémentaire incorporé. Il faut garder à l’esprit que ce procédé n’est pas infaillible et qu’il existe un taux d’erreur par nucléotide pouvant aller jusqu’à 1 %. Chaque région est lue un certain nombre de fois, ce qui correspond à la profondeur (donnée en nombre de lectures ou « X ») et qui est fonction à la fois de la quantité de fragments de librairie pouvant être lus sur la puce de séquençage, de la taille du panel et du nombre d’échantillons multiplexés. Cette profondeur conditionne la sensibilité de séquençage et doit être généralement supérieure à 1 000 pour pouvoir détecter des mutations sous-clonales représentant 1 à 2 % des cellules.
Les fichiers générés par le séquenceur font alors l’objet d’un traitement bio-informatique qui comporte les étapes suivantes : démultiplexage (pour attribuer chaque séquence au bon patient), alignement des séquences sur le génome de référence, appel de variants (pour identifier les variations de séquence par rapport au génome de référence) et annotation des variants par l’utilisation de données publiques. Chacune de ces étapes nécessite une optimisation pour éviter les faux positifs ou les faux négatifs. Le biologiste doit ensuite étudier chaque variant pour ne retenir que ceux qui ont une pertinence clinique. Le compte-rendu comporte une liste de variants identifiés par le nom du gène, le numéro d’ARN messager (NM), la nature des variants sur la séquence nucléotidique (génomique, précédée de g., ou sur l’ADN complémentaire rétrotranscrit à partir d'un ARN messager, précédé de c.) et protéique (précédée de p.) et la fréquence allélique du variant (VAF par utilisation de l’acronyme anglais variant allele frequency) qui correspond à la proportion de lectures du variant par rapport à celles de la référence et qui est un reflet de la taille du clone tumoral portant la mutation. Chaque variant est assorti d’une prédiction d’effet selon des recommandations internationales (variant pathogène, probablement pathogène, de signification indéterminée, probablement bénin ou bénin).5,6 Enfin, une interprétation du profil moléculaire dans sa globalité est indispensable pour guider la décision clinique.
Des indicateurs qualité sont intégrés aux différentes étapes et permettent ainsi d’assurer la qualité du résultat rendu (concentration et taille des librairies, paramètres de séquençage, profondeurs de lecture, etc.), qui est également contrôlée par des campagnes d’évaluation externes de la qualité.7
Nombreuses indications du SHD dans les hémopathies malignes
Sous l’égide de la Société française d’hématologie, le Groupe des biologistes moléculaires des hémopathies malignes (GBMHM) a proposé une synthèse des indications du SHD dans les principales hémopathies malignes, disponible sur le site www.gbmhm.fr (tableau).
Syndromes myélodysplasiques et myélodysplasiques/myéloprolifératifs
Le diagnostic des syndromes myélodysplasiques repose sur une expertise cytologique délicate. En l’absence de certitude diagnostique, le SHD permet de démontrer la clonalité de l’hématopoïèse. Celle-ci doit cependant être interprétée avec précaution, étant donné la prévalence importante de l’hématopoïèse clonale dans la population générale saine âgée de plus de 50 à 60 ans. L’hématopoïèse clonale correspond à la détection de mutations acquises dans des gènes impliqués dans la leucémogenèse chez des sujets sains, sans hémopathie.8
Le SHD permet également de détecter un éventuel syndrome de prédisposition génétique, ce qui peut être important lorsqu’une greffe intrafamiliale est possible. De plus, le séquençage est devenu indispensable pour préciser le pronostic de ces patients avec le score IPSS-M (International prognostis scoring system–molecular, qui inclut des paramètres de l’hémogramme, des données cytogénétiques et des données moléculaires) et donc adapter la prise en charge thérapeutique.9 Dans les syndromes myélodysplasiques/myéloprolifératifs, le séquençage a également un intérêt diagnostique pour certaines entités rares ou pour préciser le pronostic, notamment dans la leucémie myélomonocytaire chronique. Et il peut permettre d’identifier des marqueurs théranostiques * (SF3B1 et luspatercept, par exemple).
Leucémies aiguës myéloïdes
Le diagnostic positif de leucémie aiguë myéloïde (LAM) ne repose pas sur l’analyse moléculaire sauf dans certaines formes cliniques comme les sarcomes myéloïdes. Cependant, le SHD est désormais nécessaire pour établir le diagnostic de certaines entités selon la classification de l’Organisation mondiale de la santé (OMS), pour déterminer le pronostic et définir le traitement, selon les classifications de l’European Leukemia Net.10 Certaines entités frontières entre syndromes myélodysplasiques et LAM sont maintenant mieux caractérisées par le séquençage que par le pourcentage de blastes, qui est le critère diagnostique classique des LAM (par exemple, les maladies avec mutation du gène TP53). De plus, le séquençage a une place pour identifier des marqueurs théranostiques (IDH1, IDH2, FLT3) en complément des techniques usuelles et pour préciser les indications de greffe de cellules souches hématopoïétiques. Concernant le sous-type particulier des leucémies aiguës promyélocytaires (LAM3), vu l’efficacité des traitements spécifiques de cette entité, il n’y a pas d’indication au séquençage haut débit dans ces maladies.
Myélofibrose primitive, thrombocytémie essentielle et polyglobulie de Vaquez
Le diagnostic de ces entités repose actuellement sur les analyses morphologiques et moléculaires (recherche de mutations des gènes JAK2, CALR, MPL). Dans les cas où ces gènes ne sont pas mutés (dits triple négatifs), le SHD est indispensable pour rechercher une preuve de clonalité de l’hématopoïèse qui, là encore, doit être interprétée avec précaution au vu de la prévalence de l’hématopoïèse clonale. Le séquençage peut également être indiqué en cas de présentation clinico-biologique atypique afin de ne pas méconnaître une forme frontière entre syndrome myélodysplasique et myéloprolifératif, le plus souvent associée à un profil moléculaire plus complexe et à un pronostic plus réservé.
De plus, certaines mutations (par exemple, celles concernant le gène TP53) sont associées à un pronostic défavorable et doivent donc être recherchées chez les patients avec une myélofibrose primitive ou secondaire éligibles à un traitement intensif par allogreffe de cellules souches hématopoïétiques.
Leucémie myéloïde chronique
L’impact de la présence de mutations drivers impliquées dans les pathologies myéloïdes chez les patients nouvellement diagnostiqués pour une leucémie myéloïde chronique est en cours d’étude. Aussi, en dehors de cas particuliers ou d’études cliniques, il n’y a pour l’instant pas d’indication à réaliser ce type d'analyse dans cette situation.
Autres syndromes myéloprolifératifs
Concernant les syndromes myéloprolifératifs rares tels que la leucémie chronique à polynucléaires neutrophiles ou les hémopathies myéloïdes à éosinophiles liées à des réarrangements de PDGFRA ou PDGFRB ou FGFR1, le diagnostic repose sur des anomalies moléculaires recherchées par séquençage à haut débit : mutations de CSF3R pour la leucémie chronique à neutrophiles, et réarrangements des gènes PDGFRA ou PDGFRB ou FGFR1 (détectables par séquençage de l’ARN) pour les hémopathies myéloïdes à éosinophiles.
Leucémie lymphoïde chronique
Le diagnostic positif ne nécessite pas de SHD dans la plupart des cas, sauf pour préciser le diagnostic par rapport aux autres syndromes lymphoprolifératifs si l’analyse cytologique et immunophénotypique est atypique. Le choix thérapeutique de première ligne dans la leucémie lymphoïde chronique (LLC) est fondé sur la présence d’anomalies de TP53 (délétion 17p et/ou mutations). Les anomalies de TP53 entraînent en effet une résistance à l’immunochimiothérapie et constituent donc une indication au traitement par les inhibiteurs de BTK en première ligne.
Les mutations dont la VAF est inférieure à 10 % (sous-clonales) ayant le même impact défavorable que les mutations dites clonales (VAF > 10 %),11 il est indispensable d'avoir recours au SHD pour les rechercher avec un panel ciblé, selon le référentiel français proposé par le groupe FILO (French Innovative Leukemia Organization).
Chez les patients en rechute ou réfractaires, la recherche de mutations TP53 est nécessaire avant toute nouvelle ligne thérapeutique car leur fréquence augmente avec l’évolution de la maladie et elles sont fréquemment associées à la transformation en lymphome agressif de Richter. La recherche de mutations de résistance aux thérapies ciblées (BTK, PLCG2 pour les inhibiteurs de BTK, BCL2 pour le vénétoclax) peut permettre d’expliquer une évolution sous thérapie ciblée.
Autres hémopathies lymphoïdes matures
Certains syndromes lymphoprolifératifs sont associés à des mutations assez spécifiques, telles que celle du gène BRAF dans la leucémie à tricholeucocytes, ou celle de MYD88 dans la maladie de Waldenström. Leur recherche peut donc apporter une aide au diagnostic dans les situations complexes. Dans les lymphomes, le profil mutationnel prend de plus en plus d’importance pour classifier les lymphomes B diffus à grandes cellules ou pour aider au diagnostic complexe des lymphomes T, par exemple. Dans les hémopathies lymphoïdes, la place de l’analyse moléculaire est toutefois moins développée que dans les hémopathies myéloïdes, sans doute du fait de l’efficacité remarquable de traitements ciblant non pas un génotype mais un phénotype comme l’expression du CD20 (rituximab, anticorps bispécifiques) et/ou du CD19 (CAR-T cells).
Perspectives d’application du SDH
Les dix années qui viennent de s’écouler ont permis de diffuser et de standardiser l’analyse d’un panel de gènes pour le diagnostic des hémopathies malignes. En se projetant vers l’avenir, les applications du SHD sont multiples.
Tout d’abord dans le suivi de la maladie résiduelle, jusqu’alors limité à certaines cibles par des techniques de PCR (polymerase chain reaction) quantitative, le SHD présente l’atout de pouvoir suivre l’ensemble des mutations du diagnostic mais aussi d’identifier de potentiels clones émergents. Il manque cependant actuellement de standardisation à la fois technique (sensibilité, analyse bio-informatique) et clinique (impact de la persistance de mutation…) dans cette indication.13
Ensuite, en utilisant de nouveaux matériels biologiques : l’analyse de l’ADN tumoral circulant a démontré son intérêt pour le diagnostic des lymphomes et l’évaluation de la réponse thérapeutique ; reste maintenant à développer cette technologie complexe pour l’offrir à tous les patients.
Un autre développement repose sur l’analyse d’autres paramètres sur les cellules tumorales :
- le séquençage des ARN messagers est déjà mis en œuvre par certains centres, ainsi que par les laboratoires du Plan France médecine génomique 2025 ; cette approche permet la détection de transcrits de fusion oncogéniques et la détection de sous-groupes transcriptomiques qui peuvent avoir un impact clinique, comme par exemple les leucémies aiguës lymphoblastiques B de type Ph-like ;
- les techniques d’analyse du profil épigénétique (modifications structurelles de la chromatine ne touchant pas la séquence de l’ADN) apparaissent également prometteuses : ATAC-sequencing, analyse des profils de méthylation par séquençage après prétraitement au bisulfite ou par l’utilisation de nouvelles générations de séquenceurs.
Enfin, le monde de la recherche est déjà passé de l’analyse de tissus (grand nombre de cellules ou « bulk ») à l’analyse de cellules uniques, soit par séquençage de l’ARN, soit par séquençage de l’ADN. Ces approches nécessitent de solides expertises bio-informatiques et leur pertinence pour la clinique est encore largement hypothétique, mais il est probable que des efforts seront fournis pour explorer cette stratégie.
Au-delà des développements technologiques, l’avenir de la biologie moléculaire passe aussi par l’intégration des données et l’identification de modèles simplifiés d’intérêt clinique pour lesquels les approches d’intelligence artificielle ont démontré leurs performances. Le succès de ces évolutions reposera sur la qualité des données générées, sur la capacité des spécialistes à les partager et sur le développement de nouveaux métiers du diagnostic intégrant ingénieurs, mathématiciens et informaticiens. Tout cela a un coût et il faut souligner l’importance des études médico-économiques qui permettent de guider au mieux le financement de ces actes.14 Il faut espérer que les investissements à venir seront à la hauteur des enjeux pour continuer à développer le SHD, dans l’intérêt des patients.
2. Goodwin S, McPherson JD, McCombie WR. Coming of age: Ten years of next-generation sequencing technologies. Nat Rev Genet 2016;17(6):333-51.
3. Sujobert P, Le Bris Y, de Leval L, et al. The need for a consensus next-generation sequencing panel for mature lymphoid malignancies: HemaSphere 2018;3(1):e169.
4. Alcazer V, Sujobert P. Panel informativity optimizer: An R package to improve cancer next-generation sequencing panel informativity. J Mol Diagn 2022;24(6):697-709.
5. Li MM, Datto M, Duncavage EJ, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 2017;19(1):4-23.
6. Koeppel F, Muller E, Harle A, et al. Standardisation of pathogenicity classification for somatic alterations in solid tumours and haematologic malignancies. Eur J Cancer 2021;159:1-15.
7. Alary AS, Maute C, Kosmider O, et al. Improvement of standardization of molecular analyses in hematology: The 10-year GBMHM French experience. Hemasphere 2021;5(12):e658.
8. Jaiswal S, Ebert BL. Clonal hematopoiesis in human aging and disease. Science 2019;366(6465):eaan4673.
9. Bernard E, Tuechler H, Greenberg PL, et al. Molecular international prognostic scoring system for myelodysplastic syndromes. NEJM Evid 2022;1(7):EVIDoa220008.
10. Döhner H, Wei AH, Appelbaum FR, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022;140(12):1345-77.
11. Rossi D, Khiabanian H, Spina V, et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood 2014;123(14):2139-47.
12. Malcikova J, Pavlova S, Barbara KV, et al. Low-burden TP53 mutations in CLL: Clinical impact and clonal evolution within the context of different treatment options. Blood 2021;138(25):2670-85.
13. Hirsch P, Tang R, Abermil N, et al. Precision and prognostic value of clone-specific minimal residual disease in acute myeloid leukemia. Haematologica 2017;102(7):1227-37.
14. Darlington M, Sujobert P, Kosmider O, et al. Targeted high-throughput sequencing for hematological malignancies: A GBMHM survey of practice and cost evaluation in France. HemaSphere 2023;7(9):e943.