L’hémoglobinurie paroxystique nocturne (HPN), ou maladie de Marchiafava-Micheli, a initialement été décrite à la fin du XIXe siècle chez les patients présentant une asthénie associée à une hémolyse intravasculaire et à une hémoglobinurie. Elle touche préférentiellement les jeunes adultes (âge médian de 30 ans au diagnostic), avec une prépondérance féminine, et reste très rare chez les enfants de moins de 15 ans.
L’HPN est une maladie rare (1/70 000 en Europe), clonale et acquise de la cellule souche hématopoïétique, liée à un défaut d’ancrage du glycosylphosphatidylinositol (GPI). Il en résulte une hypersensibilité à l’action lytique du complément et une hémolyse corpusculaire, associée à une part d’aplasie médullaire et à un risque accru de thromboses.
Physiopathologie
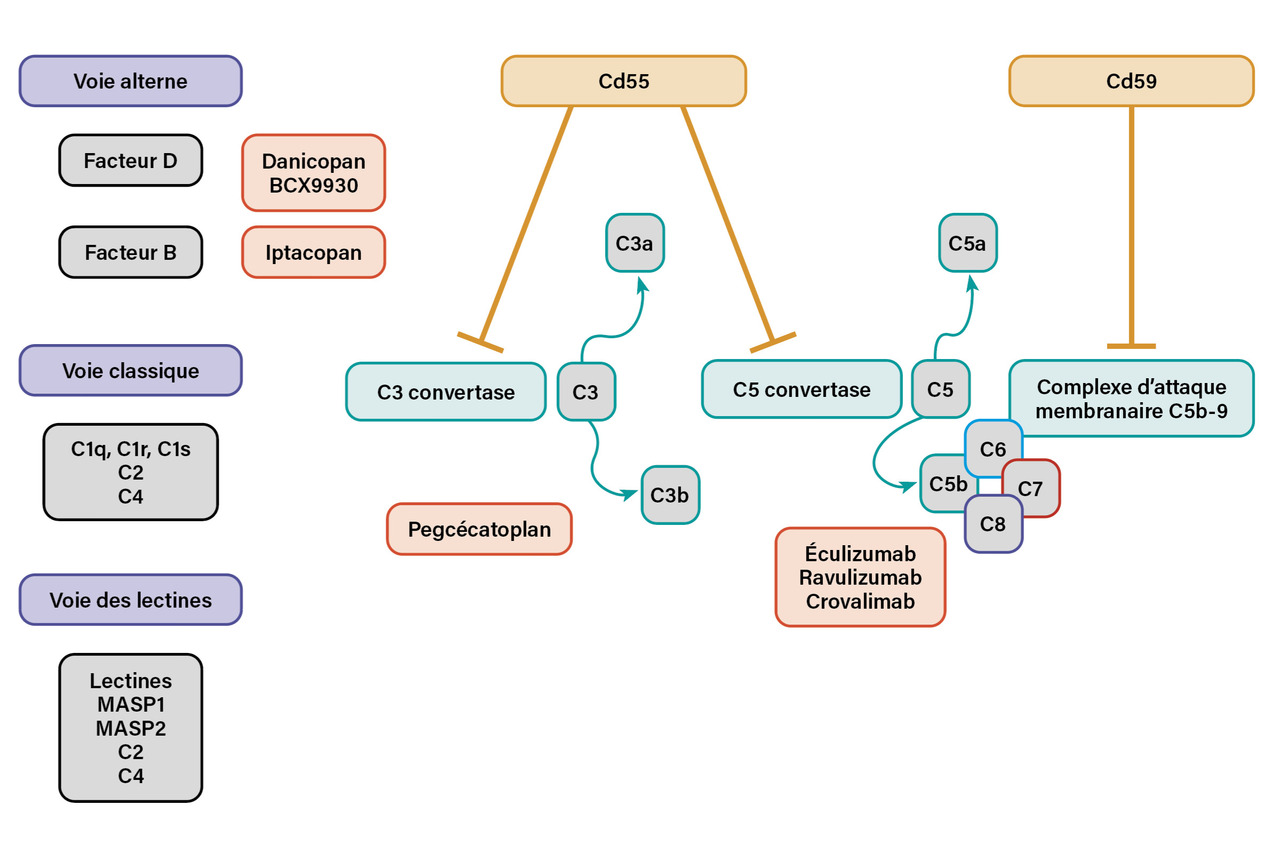
L’HPN est due à une mutation somatique acquise du gène PIGA, situé sur le chromosome X, codant pour une glycosyltransférase permettant la synthèse du GPI. Il s’agit d’une mutation par perte de fonction qui induit notamment un défaut d’expression de protéines inhibitrices du complément GPI-ancrées à la surface des cellules hématopoïétiques : le Cd55 (ou decay-accelerating factor [DAF]) inhibant les C3 et C5 convertases et le Cd59 (ou membrane inhibitor of reactive lysis [MIRL]) qui inhibe l’assemblage final du complexe d’attaque membranaire.1 Le déficit de ces deux protéines explique une sensibilité accrue à l’activité lytique du complément, induisant une hémolyse intravasculaire.
Signes cliniques et biologiques
Les signes cliniques associent à l’hémolyse une hypoplasie médullaire avec un degré de sévérité variable, des thromboses (cérébrales, splanchniques, hépatiques, abdominales, dermiques) et des signes cliniques de dystonie. Les différents signes cliniques et biologiques sont décrits dans le tableau 1.
Diagnostic biologique
Une fois le diagnostic suspecté, il est confirmé par cytométrie en flux couplée à l’aérolysine (technique FLAER), réalisée sur les monocytes et polynucléaires neutrophiles et permettant de détecter un clone HPN avec une sensibilité de 0,01 %. Le résultat obtenu est exprimé en pourcentage de cellules négatives (proportion relative du clone dans la population étudiée) ou en moyenne d’intensité de fluorescence.2 - 4 Il est important d’étudier le clone sur deux populations cellulaires différentes avec deux anticorps reconnaissant deux molécules GPI différentes. En effet, si celui-ci n’est effectué que sur les globules rouges, le résultat peut être sous-estimé du fait de la destruction des hématies, de même en cas de transfusions. L’hémolyse survient en général lorsque le clone est supérieur à 20 %.
La prise en charge initiale requiert également un hémogramme, un bilan d’hémolyse, un bilan martial (pertes urinaires de fer via l’hémoglobinurie), un myélogramme avec cytogénétique.
Formes cliniques et complications
Plusieurs entités cliniques sont classiquement décrites : l’HPN pure/de novo, la forme aplasie/HPN et la forme infraclinique (tableau 2) :3
- l’entité HPN pure, classique, est la plus fréquente, notamment chez les patients jeunes, et se caractérise par une hémolyse corpusculaire acquise, avec une hémoglobinurie matinale (20 % des patients). L’anémie peut être accompagnée de cytopénies modérées (polynucléaires neutrophiles [PNN] > 1,5 G/L, plaquettes > 120 G/L) ;
- la présentation peut être celle d’une aplasie médullaire dominée par les cytopénies. Les formes aplasie/HPN ont le plus souvent un clone HPN modéré, associé à des cytopénies (PNN < 1 G/L, plaquettes < 80 G/L, hémoglobine < 10 g/dL) ;
- les formes infracliniques ne sont pas associées à des signes cliniques ou biologiques d’hémolyse et ont une proportion de clones inférieure à 1 %. Ces formes seraient associées à une meilleure réponse au traitement immunosuppresseur dans le cadre des aplasies médullaires.
Enfin, certaines hémopathies myéloïdes (myélodysplasie notamment) peuvent être associées à une HPN.5
D’un point de vue clinique, une des manifestations fréquentes conduisant au diagnostic est la thrombose inaugurale (10 %, incidence cumulée de 40 % à dix ans), qui constitue la première cause de décès.6 Elle peut également survenir sous inhibiteur du complément en cas de blocage insuffisant. Il convient ainsi de rechercher une HPN en cas de thromboses non expliquées, atypiques (syndrome de Budd-Chiari, thrombose veineuse cérébrale), associées à des cytopénies ou en présence d’une hémolyse avec un test de Coombs négatif. La majorité des thromboses sont veineuses (85 %) et 15 % sont artérielles. Leur survenue multiplie le risque de décès par un facteur de 5 à 10.
Environ 15 % des patients présentent également des complications infectieuses (du tractus respiratoire, gastro-intestinales), qui sont la deuxième cause de décès.
En fonction des complications décrites ci-après, il convient également d’effectuer une recherche de clone HPN.
Sur le plan biologique, environ un tiers des patients se présentent avec une anémie isolée et 40 % avec une pancytopénie à moelle riche.
Durant l’évolution, à dix ans, 20 % des patients ayant une forme classique développent une aplasie médullaire.7
L’évolution clonale avec développement d’un syndrome myélodysplasique ou d’une leucémie aiguë reste rare (à dix ans 5 % et 2,5 % respectivement).
Prise en charge thérapeutique
Traitement non spécifique
Comme dans tout contexte d’hémolyse chronique, une substitution par acide folique au long cours est recommandée. Pour les femmes en âge de procréer, il convient de prescrire une contraception sans risque thrombogène. Une anticoagulation au long cours doit être poursuivie chez les patients avec un antécédent de thrombose.
Traitement spécifique
Un traitement par inhibiteur du complément est indiqué en cas de signes cliniques d’hémolyse, de complications (thromboses), de grossesse et/ou de besoins transfusionnels. Une prise en charge en affection de longue durée doit être demandée.
En première ligne, les inhibiteurs de la voie terminale (inhibiteurs de C5) sont utilisés (figure) ; ils bloquent le complexe d’attaque membranaire. L’éculizumab est le premier anticorps monoclonal humanisé à avoir été développé ;8 - 10 il a obtenu une autorisation de mise sur le marché (AMM) en 2007, suivi par le ravulizumab en 2019.11,12 Ces deux molécules se lient au C5 et préviennent son clivage en C5a pro-inflammatoire et en C5b, inhibant la formation du complexe d’attaque membranaire.
La perfusion intraveineuse d’éculizumab est effectuée à un rythme hebdomadaire les quatre premières semaines à la posologie de 600 mg par perfusion, puis de 900 mg toutes les deux semaines. Un décalage maximal de la perfusion de quarante-huit heures peut être toléré.
Après une dose de charge adaptée au poids à deux semaines, le ravulizumab est administré toutes les huit semaines par voie intraveineuse avec une posologie adaptée au poids (décalage possible de sept jours).
Un autre inhibiteur de C5, le crovalimab, s’administre mensuellement par voie sous-cutanée .13 Ce traitement doit être poursuivi à vie.
L’utilisation de bloqueurs du complément expose à un sur-risque d’infections à méningocoques. La vaccination contre les méningocoques B (avec rappel à un mois) et A, C, W, Y doit ainsi précéder l’initiation du traitement lorsque c’est possible. Les deux vaccins nécessitent ensuite un rappel tous les trois ans. Une antibioprophylaxie contre les infections à méningocoques doit être poursuivie tout au long du traitement, car un risque persiste malgré la vaccination. Les vaccinations contre les pneumocoques et Haemophilus influenzae sont également recommandées, ainsi que celle contre la grippe saisonnière et le Covid- 19, du fait d’un risque accru de poussée hémolytique.
Ce traitement permet de réduire les besoins transfusionnels, l’anémie et améliore la qualité de vie. Soixante pour cent des patients deviennent transfusion-indépendants, avec une quasi-normalisation des LDH, mais la plupart (80 %) conservent une anémie avec une hémoglobine (Hg) inférieure à 12 g/dL, une réticulocytose avec hyperbilirubinémie via une hémolyse extravasculaire14 ou un blocage insuffisant du complément.
Nouvelles perspectives thérapeutiques
Il peut persister, chez les deux tiers des patients, une hémolyse extravasculaire via l’opsonisation de C3d induite par le traitement par inhibiteurs de C5, provoquant une destruction des hématies par le système macrophagique et réticulo-endothélial.15 Pour 20 % de ces patients, des transfusions restent nécessaires.
De nouvelles thérapeutiques avec un mécanisme d’action en amont de la convertase C5 ont été développées récemment, afin d’inhiber la voie alterne du complément, et pas seulement la phase terminale d’activation au niveau du complexe d’attaque membranaire (figure).
Le pegcétacoplan est un inhibiteur proximal de la convertase C3, approuvé en 2021, qui permet de contrôler l’hémolyse intravasculaire et d’éviter l’hémolyse extravasculaire.16 Il est indiqué en cas de persistance d’une hémolyse extravasculaire sous inhibiteurs de C5 pendant plus de trois mois avec une anémie inférieure à 10,5 g/dL nécessitant des transfusions itératives (essai PEGASUS). Il s’administre par voie sous-cutanée deux fois par semaine (en association à un anti-C5 pendant quatre semaines, puis en monothérapie). L’étude a permis de montrer une médiane d’hémoglobinémie à 11,3 g/dL à seize semaines versus 8 g/dL dans le bras contrôle sous inhibiteurs de C5, avec obtention d’une indépendance transfusionnelle pour 85 % des patients versus 15 % dans le bras contrôle. En raison d’un surrisque d’infections à germes encapsulés, il est impératif que les patients soient également vaccinés contre les pneumocoques, les méningocoques et Haemophilus influenzae, et que le traitement soit initié en milieu hospitalier les deux premiers mois (risque de rebond d’hémolyse).
Plus récemment, les résultats d’un inhibiteur proximal oral sélectif du facteur B du complément ont été présentés ; il s’agit de l’iptacopan utilisé chez des patients en échec thérapeutique avec les inhibiteurs de C5 et avec persistance d’une hémolyse extravasculaire (figure). Ce médicament a obtenu une autorisation de mise sur le marché (AMM) en mai 2024 (en monothérapie orale). Une indépendance transfusionnelle est observée chez 97 % des patients recevant l’iptacopan en monothérapie et 85 % ont une élévation de l’hémoglobine basale de 2 g/dL.17
D’autres inhibiteurs proximaux sont en cours d’évaluation, notamment du facteur D ; ils s’administrent également par voie orale en association avec un inhibiteur de C5 (danicopan, ayant obtenu une AMM en avril 2024, en administration par voie orale, et en association avec le ravulizumab ou l’éculizumab)18 ou en monothérapie (BCX) [figure].19,20
Situations particulières
Certaines situations physiologiques (grossesse, notamment au troisième trimestre) ou pathologiques (contexte chirurgical, infectieux) restent à haut risque pour les patients sous traitement, augmentent la synthèse de complément et nécessitent un monitoring très régulier des symptômes et des paramètres d’hémolyse, ainsi que l’augmentation des doses d’éculizumab ou de ravulizumab, ou le rapprochement des perfusions.
Surveillance
Une surveillance de l’hémogramme doit être réalisée tous les trois mois. Un suivi annuel par le médecin référent doit être effectué, avec contrôle de l’évolution du clone HPN et réalisation d’un myélogramme avec cytogénétique pour vérifier l’absence d’évolution clonale tous les douze à dix-huit mois.
Le patient doit être éduqué vis-à-vis des situations à risque de thrombose (tabagisme, voyage en avion de plus de trois heures, immobilisation, chirurgie, grossesse) et des symptômes nécessitant une consultation en urgence (fièvre, céphalées, douleurs abdominales ou thoraciques).
Il est important que le calendrier vaccinal soit respecté.
Une maladie rare qu’il faut néanmoins savoir évoquer
L’HPN est une maladie rare à évoquer devant une anémie hémolytique sans cause corpusculaire ou immunologique identifiée, notamment dans un contexte de thrombose ou d’aplasie médullaire. Le pronostic et la qualité de vie sont nettement améliorés par l’apport des inhibiteurs terminaux et plus récemment proximaux du complément avec de nouvelles thérapeutiques en cours de développement.
2. Richards SJ, Whitby L, Cullen MJ, Dickinson AJ, Granger V, Reilly JT, et al. Development and evaluation of a stabilized whole-blood preparation as a process control material for screening of paroxysmal nocturnal hemoglobinuria by flow cytometry. Cytometry B Clin Cytom 2009;76(1):47‑55.
3. Parker C, Omine M, Richards S, Nishimura JI, Bessler M, Ware R, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood 2005;106(12):3699‑709.
4. Borowitz MJ, Craig FE, Digiuseppe JA, Illingworth AJ, Rosse W, Sutherland DR, et al. Guidelines for the diagnosis and monitoring of paroxysmal nocturnal hemoglobinuria and related disorders by flow cytometry. Cytometry B Clin Cytom 2010;78(4):211‑30.
5. Sutra Del Galy A, Willems L, D’Aveni M, Pautas C, Chantepie S, Carpentier B, et al. Haemolytic paroxysmal nocturnal haemoglobinuria in patients with myeloid neoplasms: A rare association with specific therapeutic implications. Br J Haematol 2023;201(2):e16‑20.
6. Ziakas PD, Poulou LS, Rokas GI, Bartzoudis D, Voulgarelis M. Thrombosis in paroxysmal nocturnal hemoglobinuria: sites, risks, outcome. An overview. J Thromb Haemost 2007;5(3):642‑5.
7. de Latour RP, Mary JY, Salanoubat C, Terriou L, Etienne G, Mohty M, et al. Paroxysmal nocturnal hemoglobinuria: Natural history of disease subcategories. Blood 2008;112(8):3099‑106.
8. Hillmen P, Muus P, Dührsen U, Risitano AM, Schubert J, Luzzatto L, et al. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood 2007;110(12):4123‑8.
9. Hillmen P, Hall C, Marsh JCW, Elebute M, Bombara MP, Petro BE, et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med 2004;350(6):552‑9.
10. Hillmen P, Young NS, Schubert J, Brodsky RA, Socié G, Muus P, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med 2006;355(12):1233‑43.
11. Lee JW, Sicre de Fontbrune F, Wong Lee Lee L, Pessoa V, Gualandro S, Füreder W, et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: The 301 study. Blood 2019;133(6):530‑9.
12. Kulasekararaj AG, Hill A, Rottinghaus ST, Langemeijer S, Wells R, Gonzalez-Fernandez FA, et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor-experienced adult patients with PNH: The 302 study. Blood 2019;133(6):540‑9.
13. Röth A, Nishimura JI, Nagy Z, Gaàl-Weisinger J, Panse J, Yoon SS, et al. The complement C5 inhibitor crovalimab in paroxysmal nocturnal hemoglobinuria. Blood 2020;135(12):912‑20.
14. Debureaux PE, Kulasekararaj AG, Cacace F, Silva BGP, Calado RT, Barone F, et al. Categorizing hematological response to eculizumab in paroxysmal nocturnal hemoglobinuria: A multicenter real-life study. Bone Marrow Transplant 2021;56(10):2600‑2.
15. Hill A, Rother RP, Arnold L, Kelly R, Cullen MJ, Richards SJ, et al. Eculizumab prevents intravascular hemolysis in patients with paroxysmal nocturnal hemoglobinuria and unmasks low-level extravascular hemolysis occurring through C3 opsonization. Haematol 2010;95(4):567‑73.
16. Hillmen P, Szer J, Weitz I, Röth A, Höchsmann B, Panse J, et al. Pegcetacoplan versus eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med 2021;384(11):1028‑37.
17. Risitano AM, Röth A, Soret J, Frieri C, de Fontbrune FS, Marano L, et al. Addition of iptacopan, an oral factor B inhibitor, to eculizumab in patients with paroxysmal nocturnal haemoglobinuria and active haemolysis: An open-label, single-arm, phase 2, proof-of-concept trial. Lancet Haematol 2021;8(5):e344‑54.
18. Danicopan as add-on therapy to a C5 inhibitor in paroxysmal nocturnal hemoglobinuria (PNH) participants who have clinically evident extravascular hemolysis (EVH) (ALPHA). NCT Identifier NCT04469465.
19. McDonald A, Le Roux Malherbe J, Kulasekararaj A, et al. Factor D inhibition with Oral BCX9930 monotherapy leads to sustained control of hemolysis and symptoms over 48 weeks in subjects with paroxysmal nocturnal hemoglobinuria Haive to C5 inhibitors. European Hematology Association 2022.
20. Kulasekararaj A, Fureder W, Mc Donald A, et al. Factor D inhibition with oral BCX9930 leads to sustained control of hemolysis and symptoms over 48 weeks in patients with paroxysmal nocturnal hemoglobinuria inadequately controlled on C5 inhibitors. European Hematology Association 2022.