Service d’oncologie pédiatrique, Hôpital d’enfants, CHRU de Nancy, 54500 Vandœuvre-lès-Nancy, France
Particularités épidémiologiques
Les cancers de l’enfant sont rares mais représentent, en France, la première cause de mortalité par maladie après l’âge de 1 an. Leur incidence est d’environ 1 700 nouveaux cas chez les enfants âgés de moins de 15 ans et de 700 cas chez les adolescents âgés de 15 à 18 ans, soit moins de 1 % de l’ensemble des cancers.
En France, environ 1 enfant sur 500 est atteint de cancer avant l’âge de 15 ans, avec une prédominance masculine (sex-ratio : 1,2).
Types de cancers
Les leucémies associées aux lymphomes représentent environ 40 % des cancers de l’enfant et environ 60 % sont des tumeurs solides
Âge
Étiopathogénie
Le dépistage des cancers n’existe pas en raison de leur rareté ; cependant, une très faible proportion d’entre eux surviennent dans le cas de syndromes de prédisposition génétique et nécessitent un suivi spécifique de dépistage :
formes héréditaires du rétinoblastome (toutes les formes bilatérales et 10 % des formes unilatérales). Ces patients ont également un risque plus élevé que la population générale de faire un deuxième cancer (anomalie constitutionnelle du gène RB1 situé en 13q14) ;
syndrome de Li-Fraumeni (mutation constitutionnelle responsable d’une anomalie de la protéine p53 et d’un défaut d’apoptose) exposant à un risque accru de tumeurs des tissus mous, des os, de lymphomes, de tumeurs cérébrales, ou de cortico- surrénalome ;
syndrome de Beckwith-Wiedemann (mutation constitutionnelle en 11p15) exposant à un risque accru de néphroblastome et d’hépatoblastome ;
trisomie 21 associée à un risque très élevé de leucémie ;
neurofibromatoses de types 1 et 2 s’accompagnant principalement de tumeurs du système nerveux bénignes ou malignes.
D’autres syndromes génétiques de prédisposition existent, encore plus rares, mais sont à rechercher dans le cadre de consultations d’oncogénétique.
Particularités diagnostiques
Ils sont banals mais doivent être reconnus précocement afin de raccourcir au minimum le délai du diagnostic et ainsi améliorer le pronostic vital, et limiter le risque de séquelles. En dehors des cancers comportant un envahissement de la moelle osseuse, les symptômes sont souvent d’apparition et d’évolution rapide alors que l’enfant conserve un bon état général. Ils sont souvent aspécifiques
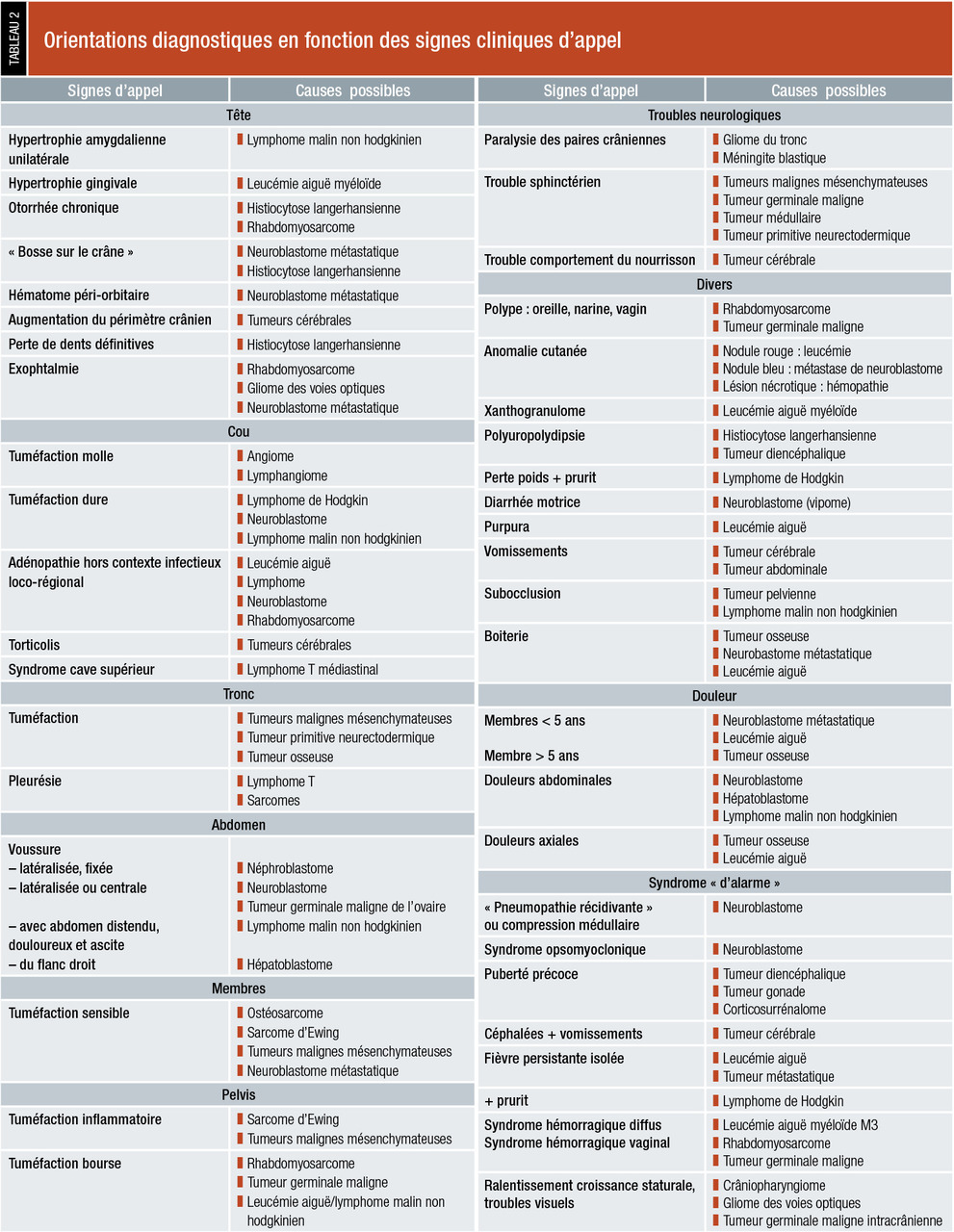
On distingue les signes directement ou indirectement en rapport avec la découverte du cancer.
Signes directement en rapport avec la découverte du cancer
Ce sont :masse abdominale, souvent de découverte fortuite qui peut être localisée en intra- ou rétropéritonéale (neuroblastome, néphroblastome, lymphome de Burkitt, principalement) ;
tuméfaction en regard d’un os ou des tissus mous au niveau des membres ou des parois du tronc en cas de tumeurs osseuses ou des parties molles (sarcomes des parties molles, ostéosarcome, sarcome d’Ewing) ;
masses ganglionnaires persistantes avec des adénopathies « froides » sans inflammation, ni douleur, ni foyer infectieux loco-régional (leucémies, lymphomes principalement) ;
masse péri- ou intra-orificielle (bouche, narines, conduit auditif, vagin, anus) ou saignement orificiel (rhabdomyosarcome principalement) ;
augmentation du volume scrotal (rhabdomyosarcome, tumeur germinale maligne) ;
reflet blanc pupillaire (« leucocorie ») ou strabisme imposant la réalisation d’un fond d’œil (rétinoblastome).
Signes révélateurs de façon indirecte
hypertension intracrânienne du fait de la tumeur elle-même et/ou de son association à une hydrocéphalie par gêne à la circulation du liquide céphalo-rachidien. Ces signes peuvent être atypiques ou insidieux (troubles visuels, troubles du comportement, augmentation trop importante du périmètre crânien). Une imagerie cérébrale doit être effectuée en cas de doute ;signes neurologiques déficitaires dont la nature dépend du siège des lésions intracrâniennes et/ou médullaires ; les convulsions sont rarement révélatrices de tumeurs cérébrales chez l’enfant.
dyspnée par compression des voies respiratoires par une tumeur médiastinale (lymphome non hodgkinien principalement) ;
obstructions respiratoires hautes ou troubles de la déglutition dus à une tumeur ORL (rhabdomyosarcome, lymphome).
Hématurie : tumeur rénale.
Fracture osseuse pathologique : tumeur primitive osseuse ou, plus rarement, métastases.
Signes ophtalmologiques : protrusion oculaire par une métastase (neuroblastome) ou une tumeur primitive orbitaire (rhabdomyosarcome, gliome du nerf optique).
Douleurs :
osseuses localisées persistantes devant faire réaliser des radiographies standard étendues des zones douloureuses et une scintigraphie osseuse au technétium (ostéosarcome, sarcome d’Ewing) ;
osseuses diffuses devant faire rechercher une atteinte de la moelle osseuse (leucémies, neuroblastome métastatique).
Autres symptômes : ce sont, par exemple, les anomalies d’émission d’urines ou de selles devant faire rechercher une tumeur abdomino-pelvienne par l’examen clinique comprenant un toucher rectal puis une échographie abdomino-pelvienne (tumeur germinale maligne, neuroblastome, rhabdomyosarcome), ou une compression médullaire (tumeur médullaire, tumeur vertébrale, neuroblastome en sablier).
Le
Les signes évocateurs d’un cancer chez l’enfant sont banals, mais leur persistance et/ou leur aggravation et/ou des douleurs nocturnes doivent faire réaliser, voire répéter, des examens complémentaires.
Diagnostic positif
Rarement, le diagnostic peut être uniquement clinique. C’est le cas du rétinoblastome dont l’aspect au fond d’œil sous anesthésie générale est caractéristique. La découverte de calcifications en échographie et au scanner permet de le conforter.
Le diagnostic peut être posé sur une convergence d’arguments cliniques et radiologiques. C’est souvent le cas du néphroblastome dont le traitement par chimiothérapie préopératoire peut être initié sans confirmation histologique si l’âge au diagnostic se situe entre 6 mois et 5 ans, si le tableau clinique et radiologique (masse intrarénale) est typique, et alors que les taux des catécholamines urinaires sont normaux.
Le diagnostic peut être posé sur une convergence d’arguments cliniques et biologiques. Dans la plupart des leucémies de l’enfant, le diagnostic est établi par l’analyse de l’hémogramme et du myélogramme. Le diagnostic peut être également posé sur la convergence d’arguments cliniques, radiologiques et de marqueurs biologiques, par exemple :
neuroblastome : élévation des taux des catécholamines urinaires (beaucoup plus fréquent chez l’enfant que le phéochromocytome), et scintigraphiques spécifiques (scintigraphie à la métaiodobenzylguanidine [MIBG]) ;
tumeurs germinales malignes sécrétantes : a-fœto-protéine et ß-hCG (respectivement tumeur du sac vitellin, choriocarcinome ou les deux marqueurs dans les tumeurs mixtes) ;
hépatoblastome : a-fœto-protéine.
Dans tous les autres cas, le diagnostic doit être posé sur la convergence d’arguments cliniques, radiologiques et cytohistologiques, soit par analyse de la tumeur primitive, soit par celle des métastases, le plus souvent alors au niveau de la moelle osseuse et/ou des adénopathies.
Dès la suspicion de tumeur maligne, les modalités et la chronologie des explorations (conservation en congélation d’un fragment tumoral, analyse en biologie moléculaire) ainsi que l’indication des abords de ponction et/ou de biopsie tumorale doivent impérativement être entreprises en milieu spécialisé afin d’éviter des retards au diagnostic, des erreurs d’interprétation ou des mesures qui pourraient être préjudiciables à la prise en charge diagnostique et/ou thérapeutique (tumeur adressée en anatomo- pathologie dans du formol plutôt qu’à l’état frais, empêchant les analyses en biologie moléculaire).
Prise en charge des situations d’urgence
Certaines urgences peuvent mettre en jeu le pronostic vital et doivent être prises en charge avant même la démarche diagnostique : choc allergique, insuffisance rénale sévère, défaillance respiratoire et/ou cardiaque, sepsis, hémorragies, thromboses étendues, état de mal convulsif.
D’autres peuvent mettre en jeu le pronostic fonctionnel, comme par exemple les complications neurologiques (hypertension intracrâniennes, crise convulsive, paraplégie).
Urgences hématologiques
Ce sont :hyperleucocytose maligne (leucocytes > 100 G/L) : signes cliniques en rapport avec une leucostase pulmonaire ou neurologique (thrombose ou hémorragies), ou des troubles métaboliques du syndrome de lyse
coagulation intravasculaire disséminée (augmentation TCA et TQ, fibrinopénie, thrombopénie) liée à une hémopathie (leucémie promyélocytaire en particulier), un syndrome de lyse tumorale en cas d’hémopathie ;
neutropénie fébrile (fièvre > 38 °C x 2 à 1 heure d’intervalle ou > 38,2 °C) + polynucléaires neutrophiles < 0,5 G/L, avec le risque de choc septique.
Urgences métaboliques
Ce sont :syndrome de lyse tumorale (acidose métabolique, hyperuricémie, hyperkaliémie, hypocalcémie) principalement observé en cas de lymphome B, et de leucémie aiguë lymphoblastique (LAL) hyperleucocytaire parfois dès le diagnostic ou à l’institution du traitement ;
insuffisance rénale aiguë, secondaire au syndrome de lyse ou à une toxicité de la chimiothérapie ;
hypercalcémie (lyse osseuse ou syndrome paranéoplasique).
Urgences respiratoires
Il s'agit surtout de la dyspnée rapidement asphyxiante en rapport avec un lymphome thoracique compressif ou avec une tumeur ORL.Urgences digestives
Ce sont :occlusion intestinale par compression digestive ou rarement invagination intestinale aiguë dues à un lymphome de Burkitt ;
pancréatite aiguë secondaire à l’utilisation de L-asparaginase et/ou de corticoïdes.
Urgences neurologiques
Ce sont :hypertension intracrânienne secondaire à une tumeur cérébrale ;
compression médullaire secondaire à un neuroblastome en sablier infiltrant les trous de conjugaison ;
état de mal convulsif secondaire à une tumeur cérébrale (rarement) ou à une toxicité de la chimiothérapie (ifosfamide, méthotrexate, vincristine).
Annonce du diagnostic
Cette annonce doit être réalisée dans les conditions prévues dans le cadre du Plan cancer. Elle doit être progressive et répétée à chaque fois que le degré d’urgence le permet, et adaptée à l’âge de l’enfant.
Le dispositif d’annonce comporte :
un temps médical : annonce du diagnostic et du programme personnalisé de soin, en évoquant la possibilité de demander un deuxième avis ;
un temps de présentation du service et des possibilités d’aides sociales, psychologiques et de soins de support ;
un temps d’information du médecin traitant qui participera au suivi durant la phase de traitement et au-delà.
Particularités thérapeutiques
Le défi thérapeutique en oncologie pédiatrique est double : guérir et sans séquelle. L’âge médian étant de 6 ans, l’espérance de vie en cas de guérison est de l’ordre de 70 ans. Il est donc primordial de choisir les traitements qui limiteront au minimum les risques de séquelles. L’enfant étant un organisme en croissance, le choix d’une stratégie thérapeutique (radiothérapie ou non, p. ex.) peut varier, pour un même cancer, en fonction de l’âge.
Rôle des facteurs pronostiques
biologique (paramètres antigéniques ou génétiques tumoraux). Par exemple, l’amplification du gène N-MYC est de très mauvais pronostic dans le neuroblastome et fait changer la stratégie thérapeutique pour les formes localisées ;
radiologique : bilan d’extension loco-régional et à distance. Par exemple, en utilisant la scintigraphie à la MIBG spécifique des localisations tumorales du neuroblastome.
Adaptation des traitements
Chimiothérapie
Elle est utilisée dans plus de 80 % des cas.Son rôle est souvent majeur, y compris avant la chirurgie (chimiothérapie néo-adjuvante), afin de diminuer le risque oncologique et la morbidité des traitements locaux.
La posologie est adaptée à l’âge et le plus souvent réduite chez le nourrisson. Les doses sont proportionnellement plus élevées que chez l’adulte, notamment dans le cadre de chimiothérapies à hautes doses suivies d’autotransfusion de cellules souches hématopoïétiques, parfois administrées à plusieurs reprises.
Pour certaines formes de leucémies aiguës (rarement dès le traitement initial, surtout lors d’une rechute), une allogreffe de cellules souches hématopoïétiques permet également d’améliorer le pronostic.
Le suivi précoce et attentif des effets secondaires, en particulier hématologiques (aplasie médullaire) et digestifs (nausées, vomissements, déshydratation, dénutrition), est primordial. Leur prise en charge nécessite assez souvent une antibiothérapie parentérale en cas de neutropénie fébrile, des transfusions en culots globulaires et en plaquettes et/ou une assistance nutritionnelle entérale ou parentérale.
Chirurgie
Elle doit être réalisée par des chirurgiens pédiatres exercés à la pratique de l’oncologie pédiatrique.Elle est parfois impossible à réaliser (certaines tumeurs cérébrales de la ligne médiane, sarcomes d’Ewing envahissant une grande partie du bassin…) mais elle est le plus souvent indispensable à la guérison des tumeurs solides.
Elle est le plus souvent réalisée après une chimiothérapie de cytoréduction qui permet de réaliser une exérèse moins délabrante.
Radiothérapie
Elle est réalisée dans environ 40 % des cas.Plus que pour tout autre traitement, sa décision tient compte du risque oncologique et de séquelles tardives (troubles fonctionnels de l’organe irradié, trouble de la croissance, risque de second cancer), d’autant plus fréquentes que l’enfant est jeune au moment de l’irradiation.
Elle doit être effectuée en milieu hautement spécialisé en utilisant la technique la plus appropriée selon la localisation tumorale (protonthérapie pour les certaines tumeurs cérébrales, p. ex.).
Biothérapies
Les thérapies moléculaires ciblées sont encore à l’étude dans la plupart des cancers de l’enfant hormis pour les leucémies avec chromosome Philadelphie et/ou fusion bcr-abl pour lesquelles un traitement par antityrosines kinases a fait ses preuves, et quelques exceptionnels autres cancers (tumeurs stromales gastro-intestinales [GIST]).Suivi des enfants après la fin du traitement
Elles sont le plus souvent observées dans les 3 ans suivant le diagnostic et sont d’autant plus graves qu’elles surviennent précocement. Le suivi (clinique, imagerie, marqueurs tumoraux…) est donc rapproché (tous les 3 à 4 mois) dans les 2 à 3 premières années, puis espacé en fonction du profil évolutif de chaque type de cancer.
Séquelles
Cette recherche est fonction de la localisation de la tumeur initiale (tumeurs cérébrales, par exemple) et des traitements administrés.
Pour les chimiothérapies
Il faut retenir :dérivés du platine (cisplatine, carboplatine) : risque de surdité, d’insuffisance rénale et/ou de tubulopathie ;
anthracyclines (doxorubicine, épirubicine, daunorubicine) : risque d’insuffisance cardiaque, parfois très tardive, risque de leucémies secondaires ;
alkylants : cyclophosphamide, busulfan : risque de stérilité ; ifosfamide : risque d’insuffisance rénale et/ou de tubulopathie ;
étoposide : risque de second cancer (leucémie) ;
bléomycine : risque de fibrose pulmonaire.
Pour la radiothérapie
Les toxicités sont dépendantes des sites irradiés :gonades : insuffisance gonadique endocrine ou exocrine ;
encéphale : risque de troubles mnésiques et de la concentration, hypopituitarisme ;
os : troubles de la croissance avec déformation osseuse ;
orbite : risque de cataracte et de rétinite ;
pelvis : risque d’hypogonadisme, de cystite ou de rectite radique ;
médiastin : risque d’insuffisance cardiaque ou de cancer des glandes mammaires.
Seconds cancers
Insertion scolaire et professionnelle
Prise en charge psychologique
Conclusion
•
POINTS FORTS À RETENIR
Les cancers de l’enfant sont rares mais représentent, en France, la première cause de mortalité par maladie après l’âge de 1 an.
Leur incidence est d’environ 2 400 nouveaux cas chez les enfants et adolescents, soit moins de 1 % de l’ensemble des cancers. L’âge médian est de 6 ans.
La première cause est représentée par les leucémies.
Leur évolution est le plus souvent très rapide, et leur diagnostic est une urgence à réaliser en milieu spécialisé.
Le traitement utilise le plus souvent la chimiothérapie, la radiothérapie dans environ la moitié des cas et la chirurgie dans presque tous les cas pour les tumeurs solides.
Le taux de survie à 10 ans est de 75 % et le risque de séquelles est très important à prendre en compte dans le choix des traitements, ainsi que celui, beaucoup plus rare, de cancer secondaire.
Traitement des cancers de l’enfant : 10 points clés
1 L’utilisation néoadjuvante de la chimiothérapie est la règle pour presque tous les cancers de l’enfant.
2 La qualité de la réponse histologique à cette chimiothérapie a une grande valeur pronostique.
3 Grande variabilité de la pharmacologie des chimiothérapies et réduction de dose nécessaire chez l’enfant âgé de moins de 1 an et/ou dont le poids est inférieur à 12 kg.
4 Abandon constant des interventions mutilantes au profit d’une chirurgie conservatrice.
5 Diminution importante des indications de radiothérapie chez le jeune enfant, surtout avant 3 ans.
6 Plus des deux tiers des malades sont inclus dans des essais thérapeutiques contrôlés avec consentement informé.
7 Fréquence de la modulation et parfois de l’amplification des séquelles avec la croissance.
8 Gravité des séquelles neuropsychiques et endocriniennes chez le jeune l’enfant irradié sur l’encéphale.
9 Pour un quart des enfants, un réseau de soins palliatifs privilégiant la vie au domicile doit être organisé.
La coopération soignants-parents est le gage de la qualité de vie au cours et au-delà du traitement.
 Encadrés
Encadrés