La sclérose en plaques (SEP) est une maladie inflammatoire chronique du système nerveux central associant des mécanismes inflammatoires et neurodégénératifs complexes (encadré et fig. 1). Les progrès majeurs réalisés au cours des dernières décennies ont profondément modifié sa prise en charge, avec le développement de nombreux traitements modifiant l’évolution de la maladie, en ciblant principalement l’inflammation. Depuis l’introduction des premiers immunomodulateurs dans les années 1990, le paysage thérapeutique s’est considérablement enrichi, offrant des options de plus en plus efficaces, au prix toutefois d’une surveillance clinique, biologique et radiologique rigoureuse.
Traitements de fond
Les traitements de fond de la SEP ont beaucoup évolué au fil du temps, des premiers immunomodulateurs injectables aux anticorps monoclonaux, en passant par les immunosuppresseurs oraux. La greffe de cellules souches hématopoïétiques autologues est réservée à certains patients réfractaires aux traitements de fond conventionnels.
Immunomodulateurs par voie injectable : interféron bêta et acétate de glatiramère
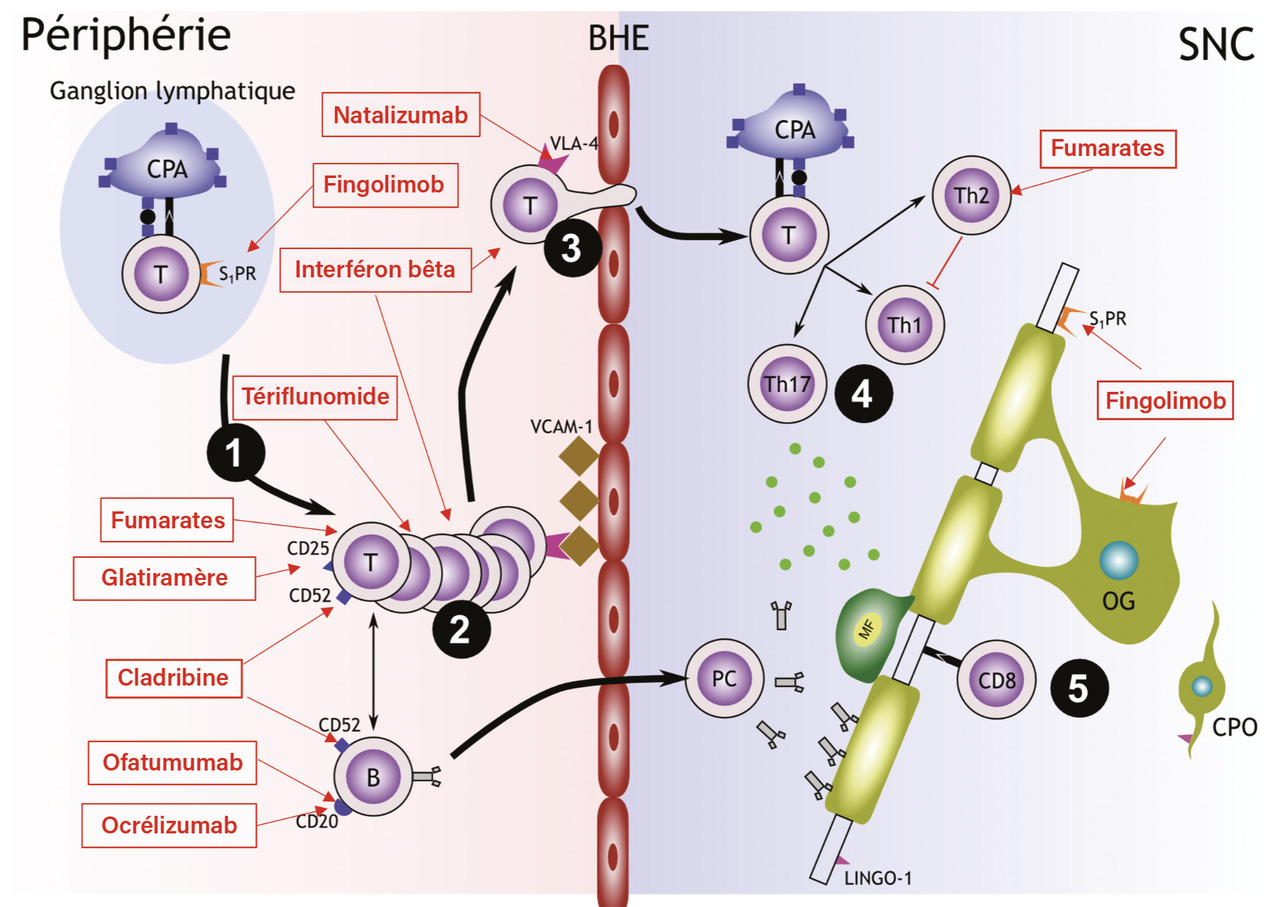
Les interférons bêta et l’acétate de glatiramère ont été les premiers traitements de fond disponibles pour la prévention des poussées de la sclérose en plaques récurrente-rémittente (RR) [tableau]. Utilisés depuis plus de trente ans, ils constituent des traitements de référence historiques de la SEP. En France, l’interféron bêta est disponible depuis le milieu des années 1990 (Betaferon en 1995, Avonex en 1997, Rebif en 1998) et l’acétate de glatiramère (Copaxone) depuis 2003 dans les formes RR de la maladie. Betaferon dispose également d’une indication dans les formes secondairement progressives avec poussées surajoutées. Les interférons bêta partagent un mécanisme d’action commun, bien que leur posologie et leur fréquence d’administration diffèrent selon les spécialités. L’interféron bêta et l’acétate de glatiramère modulent la réponse immunitaire en réduisant l’activation des lymphocytes T, en inhibant la production de cytokines pro-inflammatoires de type Th1, en favorisant une réponse Th2 anti-inflammatoire et en limitant le passage des lymphocytes autoréactifs du sang vers le système nerveux central par modulation de la perméabilité de la barrière hémato-encéphalique (fig. 2).
Ces deux traitements se distinguent toutefois par leur mode d’action spécifique. L’interféron bêta, à l’image de la protéine naturellement produite par l’organisme en réponse à une agression, se lie à des récepteurs exprimés à la surface des lymphocytes et des macrophages. L’acétate de glatiramère, copolymère de structure proche de celle de la protéine basique de la myéline (PBM), induirait quant à lui une tolérance immunitaire.
L’efficacité des interférons bêta dans les formes RR de SEP repose sur trois études pivots de phase III, conduites sur une durée de deux ans.1 - 3 Les essais comparatifs ultérieurs (QUASIMS,4 BECOME,5 REGARD6 et BEYOND7) ont montré une efficacité clinique et radiologique comparable entre interférons bêta et acétate de glatiramère. Ces traitements permettent une réduction d’environ 30 % de la fréquence des poussées et diminuent le nombre de nouvelles lésions visibles en imagerie par résonance magnétique (IRM).
Ces immunomodulateurs injectables administrés par voie sous-cutanée peuvent entraîner des réactions locales au point d’injection, telles que rougeur, induration, ecchymoses, voire plus rarement une nécrose, parfois associées à des douleurs ou un prurit.
Les interférons bêta sont fréquemment responsables d’un syndrome pseudogrippal (frissons, myalgies, céphalées, fièvre) survenant dans les heures suivant l’injection. Ces manifestations générales sont le plus souvent transitoires et s’atténuent au cours des premiers mois de traitement. En raison du risque de cytolyse hépatique ou de leucopénie, une surveillance biologique régulière est recommandée, avec un contrôle mensuel de l’hémogramme et des transaminases pendant les trois premiers mois, puis tous les trois à six mois.
L’acétate de glatiramère ne nécessite pas de surveillance biologique spécifique et n’induit pas de syndrome pseudogrippal. Des réactions locales au site d’injection sont toutefois fréquentes. La survenue de réactions systémiques transitoires à type d’oppression thoracique immédiatement après l’injection a été rapportée, sans contre-indiquer la poursuite du traitement.
Immunosuppresseurs oraux
Il en existe quatre types : tériflunomide, fumarates, inhibiteurs de la S1P et cladribine.
Tériflunomide
Le tériflunomide est un immunomodulateur oral dérivé de la léflunomide, dont le mécanisme d’action repose sur l’inhibition réversible de la dihydro-orotate déshydrogénase (DHODH), enzyme clé de la synthèse de novo des pyrimidines. Cette inhibition limite la prolifération des lymphocytes T et B activés. En réduisant la capacité de ces cellules immunocompétentes à se multiplier, le tériflunomide diminue l’activité inflammatoire et, secondairement, l’infiltration des cellules immunitaires dans le système nerveux central.
Administré par voie orale quotidienne, le tériflunomide réduit la fréquence annualisée des poussées d’environ 30 % par rapport au placebo et diminue d’environ 60 % le nombre de nouvelles lésions actives détectées à l’IRM.8 Les essais cliniques ont également montré une réduction significative, de l’ordre de 30 %, du risque d’aggravation confirmée du handicap neurologique.9 Bien que son efficacité soit modérée, ce traitement occupe une place dans la stratégie thérapeutique de première intention, aux côtés des immunomodulateurs injectables, notamment en raison de sa simplicité d’utilisation et de son mode d’administration orale.
Le tériflunomide est indiqué chez l’adulte dans les formes RR de sclérose en plaques présentant une activité modérée, en première intention, avec une surveillance clinique, biologique et radiologique régulière tout au long du traitement.
Des effets indésirables hépatiques potentiellement sévères ont été rapportés, en particulier en début de traitement, avec des élévations parfois importantes des transaminases pouvant conduire à l’arrêt du médicament. Ces risques justifient une surveillance renforcée du bilan hépatique durant les six premiers mois, puis une surveillance régulière tout au long du traitement. Le tériflunomide peut également entraîner une diminution diffuse modérée de la densité capillaire, des troubles gastro-intestinaux, une élévation de la pression artérielle (en moyenne + 2,6 mmHg pour la pression systolique et + 1,8 mmHg pour la pression diastolique) ainsi qu’une augmentation du risque d’infections respiratoires bénignes.
Concernant sa sécurité d’emploi, une vigilance particulière est requise chez les femmes en âge de procréer en raison du caractère tératogène du tériflunomide. Une contraception efficace est donc impérative pendant le traitement. En raison de sa longue demi-vie et de son élimination prolongée, un protocole d’élimination accélérée par cholestyramine ou charbon activé est requis en cas de projet de grossesse ou d’effet indésirable sévère. Les projets de grossesse doivent être anticipés, avec arrêt du traitement, mise en œuvre de la procédure d’élimination accélérée pendant onze jours et réalisation de deux contrôles biologiques à quinze jours d’intervalle confirmant l’élimination sérique du médicament. La grossesse peut alors être envisagée et la contraception interrompue.
Fumarates
Le diméthyle fumarate et le diroximel fumarate sont des immunomodulateurs oraux dont le mécanisme d’action repose principalement sur l’activation de la voie de signalisation Nrf2, impliquée dans la réponse cellulaire au stress oxydatif. Cette activation induit l’expression de gènes cytoprotecteurs et contribue à réduire l’inflammation au sein du système nerveux central. Par ailleurs, les fumarates exercent une modulation du compartiment lymphocytaire, avec une réduction préférentielle des lymphocytes T et B à phénotype pro-inflammatoire, limitant ainsi l’infiltration de cellules immunocompétentes dans le système nerveux central.
Administré par voie orale biquotidienne, le diméthyle fumarate réduit la fréquence annualisée des poussées d’environ 50 % par rapport au placebo et diminue d’environ 70 % le nombre de nouvelles lésions inflammatoires détectées à l’IRM.10,11 Les essais cliniques ont également montré une réduction significative, de l’ordre de 30 %, du risque d’aggravation confirmée du handicap neurologique.10 Ces résultats, supérieurs à ceux observés avec les interférons bêta, ont permis de positionner le diméthyle fumarate comme un traitement de fond de première intention dans les formes RR de la sclérose en plaques, combinant une efficacité clinique notable et une administration orale simple.
La prescription des fumarates est ainsi indiquée dans les formes RR de sclérose en plaques présentant une activité modérée, en première intention, avec un suivi clinique, biologique et radiologique régulier tout au long de la prise en charge.
Les patients doivent être informés de la survenue fréquente, mais généralement bénigne, de bouffées vasomotrices (« flush ») et de trou-bles gastro-intestinaux, en particulier en début de traitement, avec une tendance à l’atténuation au fil du temps. Une diminution progressive du taux de lymphocytes est observée chez environ 30 % des patients, justifiant une surveillance hématologique régulière. Dans de rares cas, une lymphopénie prolongée sévère (< 500/mm3 pendant plus de six mois) peut survenir ; dans cette situation, l’arrêt du traitement est recommandé en raison du risque accru de leuco-encéphalopathie multifocale progressive (LEMP) rapporté en association avec des taux lymphocytaires très bas. Une surveillance du taux de lymphocytes doit donc être maintenue pendant toute la durée du traitement.
Inhibiteurs de la S1P (fingolimod, ponésimod)
Le fingolimod est un modulateur des récepteurs de la sphingosine- 1 -phosphate (S1P) qui agit en induisant une séquestration des lymphocytes T et B au sein des gan-glions lymphatiques. Phosphorylé in vivo, il se lie au récepteur S1P1 exprimé à la surface des lymphocytes et agit comme un agoniste fonctionnel entraînant l’internalisation du récepteur, empêchant ainsi la sortie des lymphocytes du compartiment lymphoïde. Cette diminution du pool de lymphocytes circulants capables de migrer vers le système nerveux central permet de réduire l’inflammation auto-immune caractéristique de la SEP. Administré par voie orale quotidienne, le fingolimod réduit la fréquence annualisée des poussées d’environ 55 % par rapport à l’interféron bêta- 1a et diminue d’environ 70 % le nombre de nouvelles lésions actives détectées à l’IRM. Les essais cliniques de phase III ont également montré une réduction d’environ 30 % du risque d’aggravation confirmée du handicap neurologique.12,13 Ces résultats, supérieurs à ceux des traitements injectables de première ligne, ont conduit à positionner le fingolimod comme un traitement d’escalade dans les formes RR actives de la maladie. Chez l’adolescent, il constitue le premier traitement oral ayant démontré une efficacité significative sur la réduction des poussées et de l’activité IRM. La prescription du fingolimod est réservée chez l’adulte et l’adolescent aux formes RR actives de SEP, en seconde intention, ou en première intention chez les patients présentant une forme sévère et d’évolution rapide, définie par la survenue d’au moins deux poussées invalidantes au cours de l’année précédente associées à une activité radiologique démontrée à l’IRM cérébrale. Le fingolimod nécessite une vigilance particulière en raison de son profil de tolérance. Le principal risque initial est la survenue de troubles de la conduction cardiaque, notamment une bradycardie ou un bloc auriculoventriculaire transitoire lors de la première administration, justifiant une surveillance électrocardiographique prolongée. D’autres effets indésirables incluent un risque accru d’infections virales, en particulier herpétiques, un œdème maculaire et une élévation des enzymes hépatiques, imposant une surveillance infectieuse, ophtalmologique et biologique régulière. De rares cas de LEMP ont été rapportés, bien qu’ils restent exceptionnels.14
Le ponésimod est un modulateur sélectif du récepteur S1P1, partageant un mécanisme d’action et une efficacité clinique globalement comparables à ceux du fingolimod. Sa sélectivité pour le récepteur S1P1 est associée à une meilleure tolérance cardiaque, permettant une initiation du traitement sans surveillance électrocardiographique prolongée lors de la première prise.
Le ponésimod peut être utilisé en première intention dans les formes actives de SEP RR, avec un profil de tolérance facilitant son utilisation en pratique clinique.
Cladribine
La cladribine est un analogue nucléosidique de l’adénosine dont le mécanisme d’action repose sur une déplétion sélective et prolongée des lymphocytes B et T. Après phosphorylation intracellulaire, la cladribine interfère avec la synthèse de l’ADN et induit l’apoptose, de manière préférentielle, au sein des populations lymphocytaires à forte activité métabolique. Les lymphocytes B et T jouant un rôle central dans la physiopathologie de la SEP, cette déplétion se traduit par une diminution durable de l’activité inflammatoire au sein du système nerveux central.
Administrée par voie orale sous forme de cures annuelles courtes, la cladribine est prescrite selon un schéma comprenant un premier cycle de cinq jours consécutifs de traitement, répété quatre semaines plus tard, puis un second cycle identique l’année suivante.
Ce traitement permet de réduire la fréquence annualisée des poussées d’environ 55 à 60 % par rapport au placebo et de diminuer de plus de 85 % le nombre de nouvelles lésions actives détectées à l’IRM. Les essais cliniques ont également montré une réduction significative (d’environ 30 %) du risque d’aggravation confirmée du handicap neurologique.15
L’efficacité prolongée obtenue après un schéma thérapeutique limité dans le temps distingue la cladribine des autres traitements de fond et en fait une option particulièrement attractive dans le cadre d’une stratégie dite de « reconstitution immune ».
En France, la prescription de cladribine est réservée aux formes RR très actives de SEP. Un suivi clinique, biologique et radiologique prolongé est recommandé avant et après chaque cure annuelle.
La cladribine nécessite une vigilance particulière en raison des effets indésirables liés à l’immunosuppression transitoire qu’elle induit. Le principal risque concerne les infections, en particulier les réactivations herpétiques, et impose une vérification préalable du statut varicelle-zona et, dans certaines situations, la mise en place d’une prophylaxie antivirale et de la vaccination. Une lymphopénie transitoire est attendue et justifie une surveillance hématologique régulière.
Anticorps monoclonaux et anthracène dione
Natalizumab et anti-CD20 sont les deux anticorps monoclonaux indiqués dans le traitement de la SEP. La mitoxantrone est utilisée pour ses propriétés immunosuppressives.
Natalizumab
Le natalizumab est un anticorps monoclonal humanisé dirigé contre l’intégrine α4. Son mécanisme d’action repose sur l’inhibition du passage des cellules immunocompétentes à travers la barrière hémato-encéphalique. L’intégrine α4β1 (VLA- 4), exprimée à la surface des lymphocytes activés et des monocytes, se lie aux molécules d’adhésion VCAM- 1 présentes sur l’endothélium vasculaire. En bloquant l’interaction VLA- 4/VCAM- 1, le natalizumab empêche l’adhésion des lymphocytes activés à l’endothélium et limite ainsi leur migration vers le système nerveux central, réduisant l’inflammation caractéristique de la SEP.
Administré par voie intraveineuse, le natalizumab diminue la fréquence annualisée des poussées d’environ 60 % et réduit d’environ 80 % le nombre de nouvelles lésions actives détectées à l’IRM.16 Les essais cliniques ont également montré, par rapport au placebo, une réduction d’environ 40 % du risque d’aggravation confirmée du handicap neurologique. Ces résultats sont nettement supérieurs à ceux observés avec les traitements immunomodulateurs injectables de première ligne, tels que les interférons bêta et l’acétate de glatiramère.
Toutefois, l’utilisation du natalizumab est limitée par le risque d’effets indésirables graves, en particulier la survenue de LEMP.17 Les premiers cas ont été décrits lors des essais de phase III, notamment chez des patients recevant une association natalizumab–interféron bêta- 1a. La LEMP est une infection opportuniste sévère liée à la réactivation du virus JC (JCV), favorisée par l’immunosuppression profonde du système nerveux central induite par le natalizumab.
En février 2024, plus de 260 000 patients avaient été exposés au natalizumab dans le monde, avec 927 cas de LEMP rapportés, survenant quasi exclusivement chez des patients ayant une sérologie JCV positive, en particulier avec un indice élevé (≥ 0,9).18
Avant toute initiation du traitement, une évaluation rigoureuse du risque de LEMP est indispensable, intégrant les principaux facteurs de risque identifiés : durée d’exposition au natalizumab, antécédents de traitement immunosuppresseur et niveau des anticorps anti-virus JC. Afin de limiter ce risque, le natalizumab est préférentiellement prescrit chez les patients ayant une sérologie JCV négative ou une sérologie positive à faible indice (≤ 0,9), avec un contrôle semestriel du titre des anticorps anti-JCV. Au-delà de deux années de traitement, la poursuite du natalizumab doit être discutée au cas par cas, après réévaluation attentive du rapport bénéfice-risque.
Le natalizumab peut être administré par perfusion intraveineuse ou par injection sous-cutanée, dans un cadre hospitalier. Il est indiqué en monothérapie chez l’adulte présentant une forme très active de sclérose en plaques évoluant par poussées, soit en seconde intention après échec des traitements de première ligne, soit en première intention dans les formes sévères caractérisées par au moins deux poussées invalidantes au cours de l’année précédente, associées à une activité radiologique démontrée à l’IRM cérébrale.
Anticorps anti-CD20
L’ocrélizumab est un anticorps monoclonal humanisé dirigé contre l’antigène CD20 exprimé à la surface des lymphocytes B, pré-B et matures. Son effet thérapeutique repose sur une déplétion sélective et prolongée de ces cellules B circulantes, via des mécanismes de cytotoxicité dépendante du complément et de cytotoxicité cellulaire dépendante des anticorps. Les lymphocytes B jouant un rôle central dans la physiopathologie de la sclérose en plaques (par leurs fonctions de présentation antigénique, de sécrétion de cytokines pro-inflammatoires et de production d’auto-anticorps), leur déplétion réduit l’activation pathologique de la réponse immune et l’inflammation au sein du système nerveux central.
Administré par voie intraveineuse, l’ocrélizumab réduit la fréquence annualisée des poussées d’environ 45 à 50 % par rapport à l’interféron bêta- 1a et diminue de plus de 90 % le nombre de nouvelles lésions inflammatoires actives détectées en IRM. Les essais cliniques OPERA ont également montré une réduction significative, de l’ordre de 40 %, du risque d’aggravation confirmée du handicap neurologique.19 Ces résultats, supérieurs à ceux des traitements de première ligne, ont positionné l’ocrélizumab comme un traitement de référence de haute efficacité dans les formes RR de la maladie.
Il s’agit par ailleurs du premier traitement ayant démontré une efficacité clinique dans la SEP primaire progressive, avec un ralentissement statistiquement significatif de la progression du handicap et une diminution de l’activité inflammatoire à l’IRM dans l’étude ORATORIO.20 Les données issues de la vie réelle suggèrent toutefois une efficacité plus hétérogène, soulignant l’importance de la sélection des patients.
L’ocrélizumab est indiqué en première intention dans les formes actives de sclérose en plaques RR chez l’adulte, en seconde intention après échec d’un traitement de première ligne ainsi que dans les formes primaires progressives précoces présentant des signes d’inflammation persistante. Un suivi clinique, biologique et radiologique strict est recommandé tout au long du traitement.
Une vigilance particulière s’impose concernant la tolérance à long terme. L’immunosuppression B prolongée expose à un risque accru d’infections, notamment respiratoires et herpétiques, et justifie une surveillance régulière des immunoglobulines sériques. De rares infections opportunistes, incluant quelques cas de LEMP, ont été rapportées chez des patients traités par anti-CD20, sans relation causale directe fermement établie avec l’ocrélizumab à ce jour. Des encéphalites à entérovirus ont également été décrites.21
L’ofatumumab est un anticorps monoclonal totalement humain dirigé contre un épitope spécifique de la molécule CD20 exprimée à la surface des lymphocytes B. Son mécanisme d’action, comparable à celui de l’ocrélizumab, entraîne une déplétion efficace des lymphocytes B et une réduction de l’activité inflammatoire au sein du système nerveux central.
Administré par voie sous-cutanée mensuelle, l’ofatumumab réduit la fréquence annualisée des poussées d’environ 50 à 60 % par rapport au tériflunomide et diminue de 80 à 90 % le nombre de nouvelles lésions inflammatoires actives détectées en IRM.22 Les essais cliniques ont également montré une réduction d’environ 30 % du risque d’aggravation confirmée du handicap neurologique. Cette efficacité, comparable à celle des autres anticorps anti-CD20, s’accompagne d’un avantage pratique majeur : une administration sous-cutanée réalisable en ambulatoire et en autonomie, sans perfusion hospitalière.
La prescription de l’ofatumumab est réservée aux formes RR actives de sclérose en plaques, en première intention chez l’adulte ou en seconde intention après échec d’un traitement de première ligne. Une surveillance clinique, biologique et radiologique continue est recommandée pendant toute la durée du traitement.
Comme pour les autres anti-CD20, la prudence est requise concernant les effets indésirables liés à l’immunosuppression prolongée. L’ofatumumab est associé à un risque accru d’infections, notamment des voies respiratoires supérieures et d’infections herpétiques, et nécessite une surveillance régulière des immunoglobulines sériques.
Mitoxantrone
La mitoxantrone est une anthracène dione initialement prescrite en cancérologie pour ses propriétés cytotoxiques.22,23 Elle possède également des propriétés immunosuppressives démontrées chez l’animal dans l’encéphalomyélite auto-immune expérimentale (EAE) et mises en évidence sur toutes les lignées impliquées dans la phase inductrice et effectrice de la démyélinisation immuno-induite.24
Dans le traitement de la SEP, trois essais randomisés contrôlés25 - 27 ont montré l’impact clinique de ce traitement sur la fréquence des poussées, la progression du handicap ainsi que son impact sur l’accumulation de lésions actives en IRM pendant la période de traitement qui variait de six à vingt-quatre mois. En France, l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) a approuvé l’utilisation de la mitoxantrone en octobre 2003, en induction mensuelle pendant six mois, à la posologie de 12 mg/m2, dans les formes agressives de SEP définies comme suit : survenue d’au moins deux poussées avec séquelles ou d’une aggravation de 2 points sur l’échelle EDSS (expanded disability status scale) dans les douze mois précédant l’initiation du traitement et présence d’au moins une lésion IRM prenant le gadolinium dans les trois mois précédant le début du traitement.
Une étude observationnelle menée dans les formes RR actives de SEP révèle, sur 100 patients, une réduction de 91 % du taux annuel de poussées dans l’année qui suit l’initiation du traitement et une réduction de 1,2 point d’EDSS pour 76 % des patients indemnes de poussées dans l’année suivant le traitement. L’activité IRM était également significativement réduite de 89 % au décours du traitement. Le bénéfice était maintenu jusqu’à cinq ans sur les poussées et jusqu’à quatre ans sur le handicap.
La mitoxantrone ne reste actuellement indiquée que dans la SEP récurrente très active associée à une invalidité d’évolution rapide lorsqu’il n’existe aucune autre option thérapeutique.
Outre le risque d’infection au cours du traitement, la toxicité cardiaque28 potentielle du médicament (réduction de la fraction d’éjection ventriculaire) limite sa durée de prescription. En outre, sa toxicité hématologique possible impose une surveillance régulière prolongée pendant cinq ans. En effet, des cas de leucémie aiguë ont été rapportés, le risque est estimé entre 1 ou 2 cas pour 1 000 patients traités.
Greffe de cellules souches
La greffe de cellules souches hématopoïétiques autologues (AHSCT) dans la SEP repose sur une déplétion profonde et transitoire des compartiments de l’immunité adaptative et innée, suivie d’une reconstitution de novo du système immunitaire. Cette stratégie thérapeutique induit une suppression durable de l’activité inflammatoire et s’accompagne d’une reconfiguration du système immunitaire, suggérant une restauration partielle de la tolérance immune.
Les données accumulées au cours des dernières années montrent que l’AHSCT est particulièrement efficace chez des patients soigneusement sélectionnés atteints de SEP RR très active, réfractaire aux traitements de fond conventionnels. L’AHSCT entraîne une suppression marquée et prolongée de l’activité inflammatoire clinique et radiologique.
En revanche, la réponse à l’AHSCT apparaît plus limitée dans la SEP secondairement progressive et reste faible dans la SEP primaire progressive, ces formes étant dominées par des mécanismes de neurodégénérescence chronique plutôt que par l’inflammation active.29
Stratégies thérapeutiques
Le choix des traitements des formes récurrentes-rémittentes et des formes progressives repose sur une évaluation individualisée. Quel qu’il soit, le traitement doit être instauré le plus précocement possible afin de limiter l’accumulation de handicaps. La prévention de l’évolution et des comorbidités est également essentielle.
Traitements des formes récurrentes-rémittentes
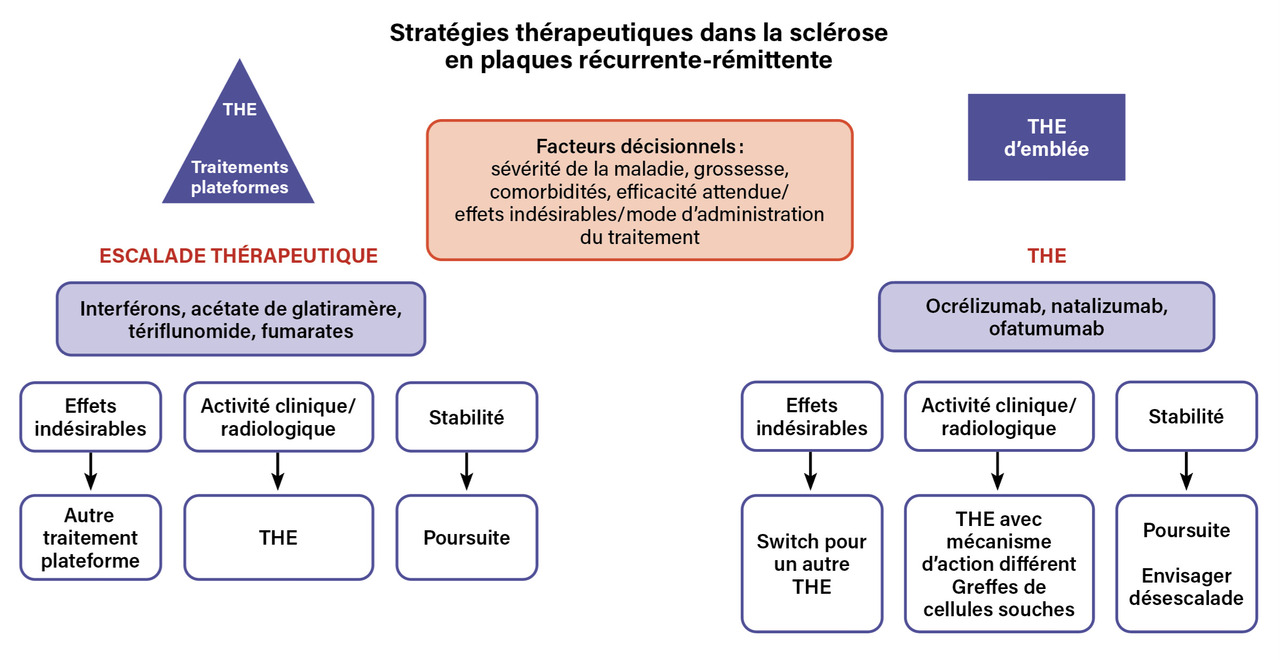
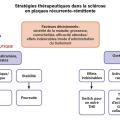
Deux grandes approches thérapeutiques coexistent actuellement dans la prise en charge de la SEP RR (fig. 3).
Escalade thérapeutique
La première, dite d’escalade thérapeutique, consiste à initier le traitement par un immunomodulateur présentant une efficacité modérée mais un profil de sécurité favorable. En cas de persistance de l’activité de la maladie manifestée par la survenue de nouvelles poussées cliniques et/ou de nouvelles lésions à l’IRM cérébrale ou médullaire, un traitement de haute efficacité (THE) est envisagé. Cette stratégie conduit classiquement à distinguer deux grandes catégories de traitement : les traitements d’efficacité modérée, dits « traitements plateformes » (interférons bêta, acétate de glatiramère, tériflunomide, fumarates), et les traitements de haute efficacité (ocrélizumab, ofatumumab, natalizumab).
Traitement de haute efficacité d’emblée
La seconde approche repose sur l’introduction d’emblée, dès le diagnostic, d’un traitement de haute efficacité, dans l’objectif de contrôler précocement l’activité inflammatoire de la maladie. Plusieurs études ont démontré l’intérêt de cette stratégie. Une étude publiée en 2021 a ainsi montré que l’initiation d’un traitement de haute efficacité dans les deux premières années suivant le début de la maladie était associée à un moindre handicap après six à dix ans d’évolution, évalué par un score EDSS plus faible, comparativement à une initiation plus tardive.30 De manière concordante, une étude rétrospective comparant les registres nationaux danois et suédois a mis en évidence qu’une stratégie privilégiant les traitements de haute efficacité en première ligne était associée à une réduction significative du risque d’aggravation confirmée du handicap à vingt-quatre semaines, ainsi qu’à une diminution du risque d’atteindre les seuils EDSS ≥ 3 et EDSS ≥ 4, par rapport à une stratégie reposant majoritairement sur des traitements d’efficacité modérée.31 Plus récemment, une étude publiée en 2025 a montré que l’initiation précoce d’un traitement de haute efficacité réduisait de plus de 75 % le risque de survenue de nouvelles lésions à l’IRM et de 70 % le risque de nouvelles poussées, comparativement à une stratégie d’escalade thérapeutique.32
Des données rétrospectives suggèrent par ailleurs que l’utilisation précoce de traitements de haute efficacité pourrait retarder la transition vers une sclérose en plaques secondairement progressive.33 Une autre étude rétrospective a également mis en évidence un impact favorable de ces traitements sur les processus neurodégénératifs, évalués par la perte de volume cérébral.34
Choix individualisé du premier traitement de fond
Le choix du premier traitement de fond repose sur une évaluation individualisée intégrant de multiples facteurs, incluant les caractéristiques cliniques de la maladie, l’activité radiologique à l’IRM, le contexte de vie du patient (notamment un éventuel projet de grossesse), les comorbidités ainsi que les propriétés propres au traitement envisagé (efficacité attendue, tolérance, modalités d’administration).
Afin d’orienter la stratégie thérapeutique, plusieurs facteurs de mauvais pronostic associés à l’aggravation du handicap doivent être pris en compte. Sur le plan clinique, il s’agit notamment de l’âge, du sexe masculin, d’un début de maladie à expression motrice ou cérébelleuse, de la présence de troubles urinaires, de poussées multiples rapprochées ou d’une récupération clinique incomplète après une poussée. Sur le plan radiologique, une charge lésionnelle élevée, l’atteinte infratentorielle ou médullaire, la présence de lésions prenant le contraste, de lésions corticales, de trous noirs persistants, de lésions chroniquement actives ainsi que d’une atrophie cérébrale ou médullaire constituent également des éléments pronostiques défavorables.
Deux messages clés doivent être soulignés : la stratégie thérapeutique doit être adaptée au profil individuel du patient dans une démarche de médecine personnalisée, et le traitement doit être instauré le plus précocement possible afin de limiter l’accumulation du handicap.
Enfin, en cas de changement de traitement motivé par un échec thérapeutique, une intolérance, un projet de grossesse ou des considérations personnelles, une vigilance particulière est requise. Certains traitements exposent, en effet, à un risque d’effet rebond à l’arrêt, notamment le fingolimod et le natalizumab. Dans ces situations, il est essentiel d’anticiper un relais thérapeutique rapide par un traitement d’efficacité équivalente, afin de prévenir la reprise de l’activité inflammatoire.
Traitements des formes progressives
La SEP de forme progressive se caractérise par une accumulation insidieuse du handicap neurologique, largement indépendante de la survenue des poussées. Elle se distingue de la forme RR, dans laquelle le handicap résulte principalement des séquelles des poussées inflammatoires. En pratique, ces deux entités ne sont pas strictement opposées : les patients s’inscrivent le plus souvent dans un continuum évolutif, avec une contribution variable des composantes inflammatoire et progressive au cours du temps.
Les formes essentiellement progressives concernent environ 15 % des patients dès le début de la maladie, définissant la SEP primaire progressive. Dans les séries historiques, antérieures à l’avènement des traitements modifiant l’évolution de la maladie, près de la moitié des patients initialement atteints d’une forme RR évoluaient secondairement vers une forme progressive, dite SEP secondairement progressive.
Les mécanismes physiopathologiques impliqués dans la progression lente et continue du handicap sont multiples et complexes. Ils reposent notamment sur une inflammation compartimentalisée du système nerveux central, distincte de l’inflammation systémique observée dans les formes RR, impliquant une activation microgliale chronique et la présence de follicules lymphocytaires B méningés. À ces phénomènes s’associent des processus de dégénérescence axonale, de perte neuronale et de dysfonction mitochondriale, contribuant à la progression irréversible du handicap.
Sur le plan thérapeutique, les anticorps anti-CD20 sont actuellement proposés chez certains patients présentant une forme progressive, sur la base des résultats de l’essai ORATORIO.20 Leur indication est renforcée en présence de signes d’activité inflammatoire, qu’elle soit clinique ou radiologique. En revanche, en l’absence d’activité inflammatoire détectable, leur impact sur la progression pure du handicap apparaît souvent limité. Dans ce contexte, un traitement d’épreuve est fréquemment proposé pour une durée d’environ deux ans, assorti d’une réévaluation régulière de l’efficacité et d’un arrêt du traitement en cas d’absence de bénéfice clinique.
Prévention de l’évolution et des comorbidités
La prévention joue un rôle central dès les premières phases de la maladie. De nombreuses données soulignent l’existence d’une fenêtre thérapeutique précoce dans la sclérose en plaques RR, au cours de laquelle une prise en charge efficace permet de réduire l’activité inflammatoire et de retarder, voire de prévenir, l’évolution vers une forme progressive.35,36
Chez les patients atteints de formes progressives, la prise en charge doit être globale et multidisciplinaire. Elle repose sur les traitements symptomatiques, la médecine physique et de réadaptation ainsi que l’adaptation au handicap et à ses conséquences fonctionnelles.
Le dépistage et le traitement des comorbidités sont essentiels, en particulier les comorbidités cardiovasculaires, qui aggravent significativement le pronostic. Le tabagisme constitue un facteur de risque bien établi, associé à une progression plus rapide du handicap et à une sévérité accrue de la SEP.37
La prévention des complications infectieuses est également primordiale, celles-ci étant plus fréquentes chez les patients présentant un handicap neurologique avancé.38 Dans ce contexte, le rôle du médecin généraliste est central pour assurer la coordination des soins, le suivi longitudinal et la continuité de cette prise en charge globale.
Centres de ressources et de compétences SEP et réunions de concertation pluridisciplinaires
Dans la prise en charge de situations complexes, ou lorsqu’une stratégie thérapeutique nécessite une évaluation approfondie du rapport bénéfice/risque, notamment en cas d’utilisation de traitements dits de seconde ligne ou hors autorisation de mise sur le marché, le recours à l’avis d’un centre de ressources et de compétences SEP (CRC SEP) est particulièrement utile. En France, 23 CRC SEP ont été labellisés en 2017 par la Direction générale de l’offre de soins (DGOS). Ces centres organisent, à l’échelle régionale, des réunions de concertation pluridisciplinaires (RCP) entre professionnels impliqués dans la prise en charge de la SEP. Les conclusions de ces réunions sont formalisées et transmises aux praticiens référents, contribuant ainsi à sécuriser les décisions thérapeutiques et à harmoniser les pratiques de prise en charge.
Traitement des poussées
Sur la base des résultats de l’Optic Neuritis Treatment Trial (ONTT),39 qui a évalué l’efficacité des corticoïdes administrés par voie intraveineuse comparativement aux corticoïdes oraux et au placebo dans le traitement des névrites optiques, la corticothérapie intraveineuse constitue le traitement de référence des poussées. Cette étude a montré un bénéfice de la voie intraveineuse sur le délai de récupération des symptômes, sans impact démontré sur le pronostic fonctionnel à long terme.
Ainsi, la méthylprednisolone administrée par voie intraveineuse à la dose de 1 g par jour pendant trois à cinq jours représente le traitement standard des poussées, conformément aux recommandations de la conférence de consensus.40
Le recours à un relais par corticoïdes oraux après la phase intraveineuse n’est pas systématique et son intérêt reste débattu.
Dans certaines situations, notamment en cas de poussées sévères ou invalidantes avec une récupération insuffisante après corticothérapie intraveineuse, le recours aux échanges plasmatiques peut être proposé.
Traitements symptomatiques
Le traitement des symptômes repose le plus souvent sur une prise en charge spécifique associant des médicaments et des techniques de rééducation.
L’apparition concomitante, au cours de l’évolution de la maladie de plusieurs symptômes impose une coordination entre les différentes spécialités impliquées dans leur prise en charge. Les consultations multidisciplinaires qui proposent au cours d’un même temps et dans un même lieu l’intervention de plusieurs spécialistes apportent une solution efficace et permettent d’améliorer la qualité de vie des patients.
Spasticité
Les médicaments utilisés pour le traitement de la spasticité sont le baclofène, le dantrolène et les benzodiazépines. Les antiépileptiques, en agissant sur la composante douloureuse de la spasticité, peuvent être utiles. À ces traitements généraux peuvent être associés des traitements locaux par injection de toxine botulinique. L’administration de baclofène par voie intrathécale au moyen d’une pompe ou des solutions chirurgicales plus radicales (neurotomie, radicotomie) peuvent être aussi proposées à des stades évolués de la maladie.
La prise en charge de la spasticité repose sur les médicaments antispastiques mais aussi sur le traitement des épines irritatives comme les infections urinaires, la constipation ou les plaies cutanées qui la favorisent et l’entretiennent.
L’immersion en eau froide peut aussi soulager les douleurs et les contractures.
Fatigue
La fatigue est un symptôme fréquent et invalidant dans la SEP. Plus de deux tiers des patients atteints de cette maladie se déclarent fatigués et la majorité d’entre eux considèrent ce symptôme comme un handicap à part entière. Dans près de 20 % des cas, elle inaugure et accompagne une poussée.
Le traitement de ce symptôme repose sur la prise en charge de toutes ses dimensions (physique, cognitive, psychosociale). La fatigue liée aux poussées est le plus souvent efficacement traitée par la méthylprednisolone par voie intraveineuse.
La fatigue chronique est, en revanche, plus difficile à traiter. Aucun traitement n’a montré son efficacité. Toutefois, la pratique d’une activité physique régulière diminuerait l’intensité de la fatigue.
Il est également utile de lutter contre l’insomnie ou les réveils nocturnes secondaires à des troubles vésico-sphinctériens.
En cas d’hypersomnie ou de somnolence diurne, un enregistrement du sommeil, à la recherche d’un syndrome d’apnées du sommeil, est nécessaire.
Troubles vésico-sphinctériens, du transit et génitaux
Le traitement des troubles urinaires a pour objectif d’assurer la continence et de maintenir une vidange vésicale complète. Un bon contrôle de la fonction vésicale améliore le confort, prévient les infections urinaires à répétition et préserve la fonction vésico-rénale.
Les troubles de la continence, en rapport avec une hyperactivité du détrusor et responsables de mictions impérieuses, de fuites urinaires ou de pollakiurie, sont efficacement améliorés par les traitements anticholinergiques : oxybutynine, trospium, solifénacine. En cas d’inefficacité de ces traitements oraux, des injections de toxine botulinique dans le détrusor peuvent être proposées.
Les troubles de la vidange vésicale, en rapport avec une hypertonie du sphincter lisse de l’urètre et responsables d’une dysurie ou d’une rétention urinaire, sont améliorés par les alphabloquants : alfuzosine, tamsulosine.
Les troubles de la continence et de la vidange vésicale peuvent coexister chez un même individu, justifiant alors l’association de différentes classes thérapeutiques. Pour adapter au mieux la thérapeutique, l’évaluation urodynamique est recommandée avant l’instauration des traitements et doit être renouvelée régulièrement au cours du suivi.
Le traitement de la constipation repose sur des mesures diététiques et les laxatifs. En cas de dyschésie anale, des suppositoires à dégagement gazeux peuvent être utiles.
Les troubles génitaux sont fréquents au cours de cette affection. Chez la femme, le traitement vise à améliorer les douleurs, les troubles de la sensibilité et la lubrification vaginale. Chez l’homme, les troubles de l’érection peuvent être traités par voie orale par sildéfanil, tadalafil, vardénafil, ou avanafil, par injections intracaverneuses d’alprostadil ou par des dispositifs locaux plus spécifiques.
Douleurs et autres symptômes
Les douleurs paroxystiques (signe de Lhermitte, névralgies faciales) sont le plus souvent soulagées par les antiépileptiques (carbamazépine, gabapentine, clonazépam).
Les paresthésies douloureuses peuvent aussi être améliorées par les antidépresseurs tricycliques ou le clonazépam.
Les douleurs articulaires secondaires au déficit moteur répondent le plus souvent aux antalgiques classiques.
D’autres symptômes nécessitent une prise en charge multidisciplinaire. Ainsi la dysarthrie, les troubles cognitifs, la dépression, les troubles respiratoires, les troubles visuels et le déficit moteur nécessitent une collaboration étroite entre neurologues, rééducateurs, urodynamiciens, ophtalmologistes, psychiatres, neuropsychologues, orthophonistes, ergothérapeutes, orthoptistes, kinésithérapeutes, psychologues et assistants sociaux.
Perspectives thérapeutiques dans la SEP
La recherche de nouveaux traitements de la SEP est très active, et de nombreux essais de phases II et III sont en cours concernant les inhibiteurs de la tyrosine kinase de Bruton, des anticorps monoclonaux, des CAR-T cells, etc.
Inhibiteurs de la tyrosine kinase de Bruton
La tyrosine kinase de Bruton (BTK) est exprimée par les lymphocytes B et les cellules myéloïdes (macrophages, cellules dendritiques, mastocytes et microglie). Contrairement aux anticorps monoclonaux, les inhibiteurs de la tyrosine kinase de Bruton (BTKi) présentent l’avantage de pouvoir pénétrer dans le système nerveux central.
Six BTKi sont actuellement en développement dans la SEP : évobrutinib, tolébrutinib, fénébrutinib, rémibrutinib, orélabrutinib, BIIB091.
Tous ont été étudiés dans les formes récurrentes-rémittentes.
À ce jour, le tolébrutinib est le seul évalué dans les formes secondairement progressives non actives. Le tolébrutinib et le fénébrutinib sont ou ont été également étudiés dans la SEP primaire progressive.
L’évobrutinib a été le premier BTKi à montrer un potentiel thérapeutique dans une étude de phase II chez des patients atteints de SEP RR avec une réduction significative de l’activité IRM.41 Cependant, les deux études de phase III (evolutionRMS 1 et 2) n’ont pas démontré de supériorité par rapport au tériflunomide sur le taux annualisé de poussées.42 Des effets indésirables graves, en particulier hépatiques, ont été signalés.
Le tolébrutinib n’a pas montré de réduction significative du taux annualisé de poussées chez des patients atteints SEP récurrente-rémittente en comparaison au tériflunomide (études de phase III GEMINI 1 et GEMINI 2).43 En revanche, l’étude HERCULES (de phase III, contrôlée versus placebo) chez des patients atteints de forme secondairement progressive (SPMS : secondary progressive multiple sclerosis) non actifs a montré une réduction de 31 % du risque de progression confirmée du handicap à six mois.44 Dans la SEP primaire progressive, des résultats récents de la phase III de l’étude PERSEUS n’ont pas montré de réduction du risque de progression confirmée du handicap à six mois. La Food and Drug Administration a donné, le 24 décembre 2025, un avis négatif à l’utilisation du tolébrutinib chez les patients atteints de SEP secondairement progressive non active.
Concernant les autres BTKi en cours d’étude, le fénébrutinib, BTKi périphérique à liaison réversible, a montré une diminution significative des lésions T1 réhaussées ainsi que des nouvelles lésions T2 dans l’étude de phase II FENopta, contre placebo.45 Les études de phase III sont en cours dans la SEP RR en comparaison au tériflunomide (FENhance 1 et 2) et dans la SEP PP (FENtrepid) comparée à l’ocrélizumab.
Le rémibrutinib, un BTKi périphérique à liaison covalente, est actuellement évalué dans deux études de phase III chez des patients SEP RR, en comparaison au tériflunomide.
L’orélabrutinib, BTKi pénétrant dans le système nerveux central, fait l’objet d’une étude de phase II contrôlée versus placebo dans la SEP RR, avec un essai dans la SEP PP en préparation.
Le BIIB091 est étudié en phase II dans la SEP RR, en monothérapie ou en association avec le diroximel fumarate.
Anticorps monoclonaux
Anti-CD40L
Le CD40, présent à la surface des lymphocytes B et des cellules dendritiques, et son ligand CD40L jouent un rôle clé dans la régulation immunitaire. Chez les patients atteints de SEP, le CD40L est surexprimé sur les lymphocytes T activés. Le fréxalimab est un anticorps monoclonal de deuxième génération anti-CD40L. Une étude de phase II a montré une réduction de 89 % des nouvelles lésions cérébrales se réhaussant en T1 au gadolinium, chez les patients SEP RR, en comparaison au placebo.46 Deux études de phase III sont en cours : une dans la SEP RR et l’autre dans la SEP secondairement progressive non active.
Foralumab par voie nasale
Le foralumab, anticorps monoclonal anti-CD3 humain, se lie au récepteur des cellules T et atténue l’inflammation en modulant la fonction des cellules T, supprimant ainsi les caractéristiques effectrices de plusieurs sous-ensembles de cellules immunitaires. Sa particularité est son mode d’administration, par voie intranasale. Cet anticorps est en cours d’étude (phase II) dans les formes de SEP secondairement progressive non active.
Vidofludimus calcium par voie orale
Le vidofludimus calcium est un inhibiteur de nouvelle génération de la dihydro-orotate déshydrogénase, la même enzyme que celle ciblée par le tériflunomide, mais avec une spécificité améliorée, offrant un meilleur profil d’effets indésirables. Après une étude de phase II concluante,47 le vidofludimus calcium administré par voie orale fait actuellement l’objet de deux essais de phase III dans les formes RR de SEP et d’un essai de phase II dans les formes progressives.
Cellules CAR-T
Les lymphocytes T à récepteur antigénique chimérique (CAR-T) sont modifiés pour reconnaître et éliminer spécifiquement les lymphocytes B autoréactifs (anti-CD19). En ciblant un large spectre de populations B (incluant les cellules naïves, mémoire, plasmablastes et les plasmocytes précoces CD19⁺), les cellules CAR-T pourraient permettre une déplétion profonde et durable des lymphocytes B. Ces cellules sont capables de franchir la barrière hémato-encéphalique et pourraient induire une rémission prolongée de la maladie, voire être potentiellement curatives.
Cependant, plusieurs risques sont associés à ces thérapies, notamment le syndrome de libération de cytokines (SLC, en anglais cytokine release syndrome, CRS) et le syndrome de neurotoxicité associée aux cellules effectrices (ICANS), pouvant aggraver la maladie. Le SLC est un syndrome inflammatoire systémique déclenché par la libération massive de cytokines après la liaison des CAR-T à leurs cellules cibles. Ses manifestations cliniques incluent fièvre, myalgies, hypoxie, hypotension artérielle et défaillance d’organes dans les cas sévères. L’ICANS résulte d’une inflammation cérébrale médiée par les cytokines engendrant des symptômes tels qu’une aphasie, des tremblements et, dans les cas sévères, des crises convulsives et une dépression respiratoire. Les données de sécurité des cellules CAR-T anti-CD19 dans la SEP sont relativement rassurantes, en comparaison aux hémopathies.
Par ailleurs, d’autres toxicités ont été décrites, chez les patients atteints de maladies hématologiques survenant dans la première semaine après traitement : syndromes hémophagocytaires, cytopénies et infections. À des stades plus tardifs, des atteintes neurologiques (trou-bles parkinsoniens, atteintes cognitives) mais aussi des atteintes tumorales T secondaires et des hypogammaglobulinémies sont rapportées.48
Plusieurs études de phase I évaluent actuellement les cellules CAR-T dans les formes récurrentes-rémittentes et progressives de la SEP. Un essai de phase II (KYSA- 7) inclut les patients atteints de SEP progressive primaire et secondaire réfractaire.49 Par ailleurs, un essai clinique de phase I est en cours, évaluant le traitement par cellules CAR-T anti-BCMA, ciblant l’antigène de maturation des lymphocytes B, chez des patients atteints de SEP progressive.
Remyélinisation et neuroprotection : deux cibles thérapeutiques ?
La neuroprotection et la remyélinisation constituent des axes majeurs de la recherche dans la SEP. De nombreux essais thérapeutiques portant sur la remyélinisation ont été menés, avec des résultats globalement modestes. Plusieurs stratégies ont été explorées, notamment la stimulation de la différenciation des précurseurs d’oligodendrocytes (antimuscariniques, agoniste du récepteur nucléaire RXR gamma), le blocage de signaux inhibiteurs (anti-LINGO- 1), des modulateurs hormonaux (œstrogènes, androgènes) ainsi que des approches reposant sur la neurostimulation et l’activité neuronale.
Les résultats peu satisfaisants des traitements potentiellement remyélinisants peuvent s’expliquer par plusieurs facteurs. Premièrement, les approches actuelles de neuroprotection ciblent principalement les neurones, alors qu’une intervention simultanée sur plusieurs types cellulaires pourrait probablement être plus efficace. Par ailleurs, il est crucial d’inclure des patients au début de l’évolution de la maladie, la capacité de remyélinisation diminuant avec l’âge et dans les formes progressives ; la fenêtre thérapeutique pour les traitements remyélinisants semble donc se situer tôt dans le cours de la maladie.
Une étude présentée lors du congrès de l’European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMs ) 2025, à Barcelone, a montré un effet léger mais significatif de l’association clémastine-metformine, avec une réduction de la latence moyenne des potentiels évoqués visuels, chez des patients atteints de SEP récurrente-rémittente avec une névrite optique chronique.
La metformine fait également l’objet d’essais comme traitement additionnel dans les formes progressives de SEP (étude MACSiMiSE-BRAIN).50
Par ailleurs, l’étude ACUITY (phase II) a montré que l’OCS- 05 réduit l’amincissement de la couche des cellules ganglionnaires et de la couche des fibres nerveuses rétiniennes, et améliore l’acuité visuelle à faible contraste lorsqu’il est utilisé chez les patients présentant une névrite optique à la phase aiguë.51
Nombreuses et récentes avancées thérapeutiques
La sclérose en plaques est une maladie complexe dans laquelle inflammation, démyélinisation et dégénérescence axonale sont étroitement intriquées. Les avancées thérapeutiques des quinze dernières années ont permis un contrôle très efficace de l’activité inflammatoire et des poussées de la maladie. Une meilleure compréhension des interactions entre ces différents mécanismes constitue désormais un enjeu majeur, afin de développer de nouvelles stratégies thérapeutiques capables de limiter la progression du handicap à long terme et de cibler plus spécifiquement la composante neurodégénérative de la sclérose en plaques.
Trois formes de sclérose en plaques
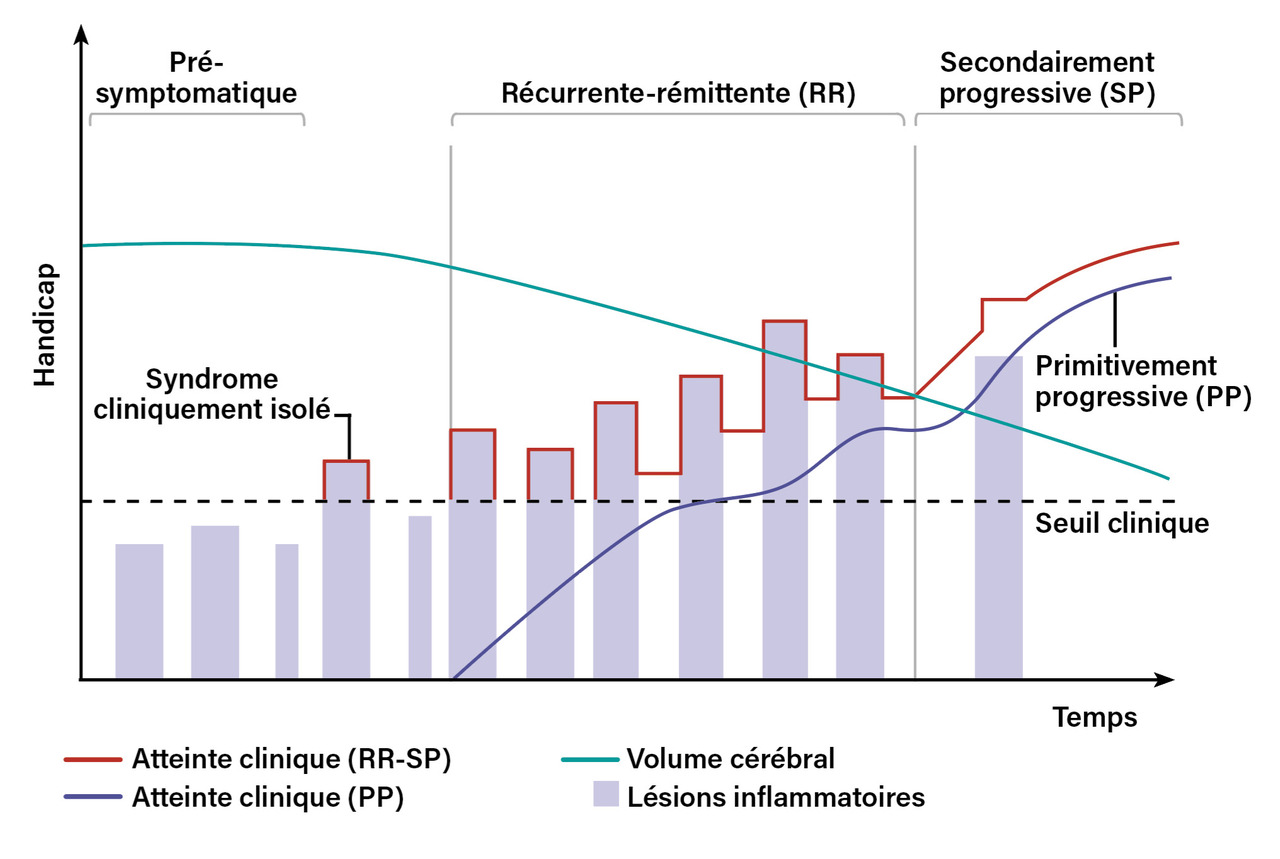
Dans 85 % des cas, la SEP se présente sous la forme dite « récurrente-rémittente » (RR), caractérisée par l’apparition de symptômes sous forme de poussées suivies de périodes de rémission plus ou moins longues (fig. 1).
Après quinze à vingt ans de maladie en moyenne, en l’absence de traitement, la plupart des patients basculent vers une forme de progression lente sans rémission, dite « secondairement progressive » (SP).
Pour 8 à 10 % des patients, en particulier ceux ayant débuté la maladie à un âge plus avancé, la SEP se présente sous forme « primaire progressive » (PP), avec une accumulation de handicaps dès le début de la maladie.
Enfin, la forme « progressive récurrente » (PR) est la moins fréquente (moins de 5 % des patients). Elle ressemble à la forme « primaire progressive » à laquelle s’ajoute l’apparition de poussées récurrentes.
2. Jacobs LD, Cookfair DL, Rudick RA, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. Ann Neurol 1996;39:285-94.
3. The PRIMS Study Group. Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing-remitting-multiple sclerosis. Lancet 1998:352(9139):1498-504.
4. Limmroth V, Malessa R, Zettl UK, et al.; QUASIMS Study Group. Quality assessment in multiple sclerosis therapy (QUASIMS): A comparison of interferon beta therapies for relapsing-remitting multiple sclerosis. J Neurol 2007;254(1):67-77.
5. Wolansky L, Cook S, Halper J, et al. Betaferon versus copaxone in MS with triple–dose gadolinium and 3-T MRI endpoints (BECOME) announcement of final primary outcome. Multiple Sclerosis Journal 2007;13:206.
6. Mikol DD, Barkhof F, Chang P, et al. Regard trial: A randomized assessor-blinded trial comparing INF beta-1a and glatiramer acetate in RR MS. Multiple sclerosis Journal 2007;13:119.
7. O’Connor P, Filippi M, Arnason B. 250 microg or 500 microg interferon beta-1b versus 20 mg glatiramer acetate in relapsing-remitting multiple sclerosis: A prospective, randomised, multicentre study. Lancet Neurol 2009;8(10):889-97.
8. O’Connor P, Wolinsky JS, Confavreux C, et al. TEMSO Trial Group. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med 2011;365(14):1293-303.
9. Confavreux C, O’Connor P, Comi G, et al.; TOWER Trial Group. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol 2014;13(3):247-56.
10. Gold R, Kappos L, Arnold DL, et al.; DEFINE Study Investigators. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med 2012;367(12):1098-107.
11. Fox RJ, Miller DH, Phillips JT et al.; CONFIRM Study Investigators. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med 2012;367(12):1087-97.
12. Cohen JA, Khatri B, Barkhof F, et al.; TRANSFORMS Study Group. Long-term (up to 4.5 years) treatment with fingolimod in multiple sclerosis: Results from the extension of the randomised TRANSFORMS study. J Neurol Neurosurg Psychiatry 2016;87(5):468-75.
13. Calabresi PA, Radue EW, Goodin D, et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol 2014;13(6):545-56.
14. Croteau D, Kim T, Chan V, et al. Progressive multifocal leukoencephalopathy associated with sphingosine-1-phosphate receptor modulators: A large case series. Mult Scler Relat Disord 2024;92:106163.
15. Giovannoni G, Comi G, Cook S et al.; CLARITY Study Group. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):416-26.
16. Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2006;354:899-910.
17. Yousry TA, Major EO, Ryschkewitsch C, et al. Evaluation for progressive multifocal leuko-encephalopathy in patients treated with natalizumab. N Engl J Med 2006;355:924-33.
18. Takahashi K, Nakahara J, Miura Y, et al. Natalizumab-associated progressive multifocal leukoencephalopathy after natalizumab extended interval dosing therapy in Japan. Neurol Neuroimmunol Neuroinflamm 2025;12(6):e200476.
19. Hauser SL, Bar-Or A, Comi G, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med 2017;376(3):221-34.
20. Banks SA, Poliakov I, Flanagan EP, et al. Enterovirus encephalitis in people with multiple sclerosis on ocrelizumab: Insights from a multicenter case series. Neurol Neuroimmunol Neuroinflamm 2026;13(2):e200534.
21. Hauser SL, Bar-Or A, Cohen JA, et al.; ASCLEPIOS I and ASCLEPIOS II Trial Groups. Ofatumumab versus teriflunomide in multiple sclerosis. N Engl J Med 2020;383(6):546-57.
22. Faulds D, Balfour JA, Chrisp P, et al. Mitoxantrone a review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in the chemotherapy of cancer. Drugs 1991;41(3):400-49.
23. Watson CM, Davison AN, Baker D, et al. Suppression of demyelination by mitoxantrone. Int J Imunopharmacol 1991;13(7):923-30.
24. Edan G, Miller D, Clanet M, et al. Therapeutic effect of mitoxantrone combined with methylprednisolone in multiple sclerosis: A randomised multicentre study of active disease using MRI and clinical criteria. J Neurol Neurosurg Psychiatry 1997;62(2):112-8.
25. Page E, Leray E, Taurin G, et al. Étude observationnelle de la mitoxantrone dans les formes rémittentes actives de SEP : suivi à long terme d’une cohorte de 100 patients consécutifs. Rev Neurol 2006;162(2):185-94.
26. Hartung HP, Gonsette R, Konig N, et al. Mitoxantrone in progressive multiple sclerosis: A placebo-controlled double blind, randomized, multicenter trial. Lancet 2002;360(9350):2018-25.
27. Ghalie RG, Edan G, Laurent M, et al. Cardiac adverse effects associated with mitoxantrone therapy in patients with MS. Neurology 2002;59(6): 909-13.
28. Protocole national de diagnostic et de soins. Greffe de cellules souches hématopoïétiques dans les maladies auto-immunes. Septembre 2022.
29. He A, Merkel B, Brown JWL, et al.; MSBase study group. Timing of high-efficacy therapy for multiple sclerosis: A retrospective observational cohort study. Lancet Neurol 2020;19(4):307-16.
30. Spelman T, Magyari M, Piehl F, et al. Treatment escalation vs immediate initiation of highly effective treatment for patients with relapsing-remitting multiple sclerosis: Data from 2 different national strategies. JAMA Neurol 2021;78(10):1197-204.
31. Norborg H, Aarseth JH, Mannseth J, et al. Effect of early highly effective treatment compared to an escalating treatment strategy in multiple sclerosis. Mult Scler Relat Disord 2025;103:106702.
32. Brown JWL, Coles A, Horakova D, et al ; MSBase Study Group. Association of initial disease-modifying therapy with later conversion to secondary progressive multiple sclerosis. JAMA 2019;321(2):175-87.
33. Uher T, Krasensky J, Malpas C, et al. Evolution of brain volume loss rates in early stages of multiple sclerosis. Neurol Neuroimmunol Neuroinflamm 2021;8(3):e979.
34. Montalban X, Hauser SL, Kappos L, et al; ORATORIO Clinical Investigators. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med 2017;376(3):209-20.
35. Tintore M, Arrambide G, Otero-Romero S, et al. The long-term outcomes of CIS patients in the Barcelona inception cohort: Looking back to recognize aggressive MS. Mult Scler 2020;26(13):1658-69.
36. Brown JWL, Coles A, Horakova D, et al.; MSBase Study Group. Association of initial disease-modifying therapy with later conversion to secondary progressive multiple sclerosis. JAMA 2019;321(2):175-87.
37. Manouchehrinia A, Tench CR, Maxted J, et al. Tobacco smoking and disability progression in multiple sclerosis: United Kingdom cohort study. Brain 2013;136(Pt 7):2298-304.
38. Brand JS, Smith KA, Piehl F, et al. Risk of serious infections in multiple sclerosis patients by disease course and disability status: Results from a Swedish register-based study. Brain Behav Immun Health 2022;22:100470.
39. Beck RW, Cleary PA, Anderson Jr MM, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. The optic Neuritis Study Group. N Engl J Med 1992;326:581-8.
40. Lubetzki C. Perspectives thérapeutiques dans la sclérose en plaques. Rev Prat 2006;56:1347-52.
41. Montalban X, Arnold DL, Weber MS et al; Evobrutinib Phase 2 Study Group. Placebo-controlled trial of an oral BTK inhibitor in multiple sclerosis. N Engl J Med 2019;380(25):2406-17.
42. Montalban X, Vermersch P, Arnold DL, et al; evolutionRMS investigators. Safety and efficacy of evobrutinib in relapsing multiple sclerosis (evolutionRMS1 and evolutionRMS2): Two multicentre, randomised, double-blind, active-controlled, phase 3 trials. Lancet Neurol 2024;23(11):1119-32.
43. Oh J, Arnold DL, Cree BAC, et al.; Tolebrutinib Phase 3 GEMINI 1 and 2 Trial Group. Tolebrutinib versus teriflunomide in relapsing multiple sclerosis. N Engl J Med 2025;392(19):1893-904.
44. Fox RJ, Bar-Or A, Traboulsee A, et al.; HERCULES Trial Group. Tolebrutinib in nonrelapsing secondary progressive multiple sclerosis. N Engl J Med 2025;392(19):1883-92.
45. Bar-Or A, Dufek M, Budincevic H, et al. Safety and efficacy of fenebrutinib in relapsing multiple sclerosis (FENopta): A multicentre, double-blind, randomised, placebo-controlled, phase 2 trial and open-label extension study. Lancet Neurol 2025;24(8):656-66.
46. Vermersch P, Granziera C, Mao-Draayer Y, et al. Inhibition of CD40L with frexalimab in multiple sclerosis. N Engl J Med 2024;390(7):589-600.
47. Fox RJ, Wiendl H, Wolf C, et al. A double-blind, randomized, placebo-controlled phase 2 trial evaluating the selective dihydroorotate dehydrogenase inhibitor vidofludimus calcium in relapsing-remitting multiple sclerosis. Ann Clin Transl Neurol 2022;9(7):977-87.
48. Samadzadeh S, Szejko N, Hamadah Y, et al. CAR T cells as novel therapeutic strategy for multiple sclerosis and other neuroimmune disorders. J Neuroinflammation 2025;23(1):35.
49. Chinas NA, Alexopoulos H. CAR T-cells meet autoimmune neurological diseases: A new dawn for therapy. Front Immunol 2025;16:1604174.
50. De Keersmaecker AV, Van Doninck E, Popescu V, et al. A merformin add-on clinical study in multiple sclerosis to evaluate brain remyelination and neurodegeneration (MACSIMISE-BRAIN): Study protocol for a multi-center randomized placebo controlled clinical trial. Front Immunol 2024;15:1362629.
51. Louapre C, Bonnin S, Mariani LL, et al. Reduction in retinal ganglion cell loss and improved low contrast visual acuity with privosegtor in acute optic neuritis: Results from a multicenter randomized placebo-controlled double-masked trial. ECTRIMS 2025;918.
 Encadrés
Encadrés