L’épileptogenèse est un processus dynamique qui modifie progressivement l’excitabilité neuronale, établit des interconnexions excitatrices et potentiellement des changements structurels avant que la première crise d’épilepsie spontanée ne se produise. Elle déclenche une vaste gamme de changements génomiques, épigénomiques et transcriptomiques, qui conduisent finalement à la transformation morphologique et fonctionnelle de circuits neuronaux spécifiques entraînant l’apparition de crises spontanées. Les dernières décennies ont permis d’élucider certains processus moléculaires et de mettre en évidence des voies de signalisation biochimiques impliquées dans la transformation proépileptique des circuits cérébraux, notamment le stress oxydatif, l’apoptose, les facteurs neuro-inflammatoires et neurotrophiques.1 Néanmoins, les mécanismes physiopathologiques exacts des crises et de l’épilepsie demeurent imparfaitement compris.
Évolution des définitions
La crise d’épilepsie est l’expression électroclinique d’une synchronisation transitoire anormale des neurones perturbant leurs réseaux de communication habituels.2
Définition des crises d’épilepsie
Classiquement, l’épilepsie était définie par la répétition d’au moins deux crises d’épilepsie. En 2014, la Ligue internationale contre l’épilepsie (International League Against Epilepsy [ILAE]) a adopté une nouvelle définition opérationnelle de l’épilepsie.2
Le diagnostic d’épilepsie peut être désormais porté si l’une des conditions suivantes est rencontrée :
Les crises provoquées restent exclues de la définition de l’épilepsie : ce sont des crises symptomatiques aiguës situationnelles liées à une situation épileptogène transitoire et réversible.4 Le délai requis pour qualifier la crise de « provoquée » après le facteur déclenchant « aigu » est au maximum de sept jours pour les causes toxiques, infectieuses ou lésionnelles (accident cardiovasculaire [AVC], traumatisme crânien, encéphalite ou intervention neurochirurgicale) et de seulement vingt-quatre heures pour les troubles métaboliques et/ou ioniques. Cela signifie concrètement l’abstention thérapeutique et la correction, si cela est possible, du facteur déclenchant. En clair, une crise d’épilepsie survenant sept jours après un AVC ou une encéphalite, ou dans les vingt-quatre heures de la constatation d’une hyponatrémie est considérée comme provoquée et ne conduit pas à l’instauration d’un traitement antiépileptique. Concernant les troubles métaboliques et/ou ioniques, des valeurs seuils ont été fixées par l’ILAE pour retenir l’imputabilité et qualifier ainsi la crise de provoquée : glycémie inférieure à 36 mg/dL (2 mmol) ou supérieure à 450 mg/dL (25 mmol) avec acidocétose, natrémie inférieure à 115 mg/dL, calcémie inférieure à 5 mg/dL ou encore magnésémie inférieure à 0,8 mg/dL.
On peut désormais porter un diagnostic d’épilepsie dès la première crise si l’on estime que le risque de survenue d’une seconde crise est élevé (supérieur à 60 %) et demeure réel sur une période prolongée (dix ans). Une revue Cochrane récente a estimé le risque de récidive après une première crise non provoquée.5 Le premier constat est que la majorité des études des taux et facteurs de récidive portent sur des périodes de deux ans, maximum cinq ans après la première crise, et que l’on dispose de très peu de données sur la récidive à dix ans (trois études seulement). Globalement, sur 46 études incluses dans la méta-analyse, le taux de récurrence des crises était de 27 % à six mois, de 36 % à un an et de 43 % à deux ans, avec des taux légèrement supérieurs de récidive chez l’enfant. D’après une méta-analyse portant sur dix études de classes I et II et sur 3 212 patients, les facteurs de risque établis de récidive, permettant ainsi de porter le diagnostic d’épilepsie dès la première crise, sont : la notion d’une agression cérébrale préexistante (par exemple un antécédent de traumatisme crânien ou d’AVC), des anomalies épileptiques sur l’électroencéphalogramme (EEG) et une imagerie cérébrale anormale.6 Un examen neurologique anormal a également été identifié comme facteur significatif de risque de récidive.
Des conséquences thérapeutiques peuvent découler de cette nouvelle définition élargie, avec des mises en route plus rapides de traitement. Les premiers (et petits) essais contrôlés randomisés de courte durée ont suggéré une réduction apparente de la récurrence des crises dans le cadre d’un traitement immédiat d’une première crise par rapport à un traitement différé. Cet avantage initial disparaît lors d’un suivi plus long avec des taux quasi identiques de rémission à trois ans et à cinq ans chez les patients dont le traitement a été initié ou retardé après une première crise non provoquée, comme l’a démontré l’étude MESS FIRST.7 Une étude récente a analysé l’effet de la définition révisée de l’épilepsie sur les décisions thérapeutiques et les taux de récurrence des crises après une première crise d’épilepsie.8 Il ressort de cette analyse de 629 patients que la proportion d’entre eux recevant un traitement antiépileptique a augmenté de manière significative, passant de 70,4 à 80,5 % (p = 0,015) à la suite de la nouvelle définition de l’épilepsie, sans changement significatif du taux de récidive (40,8 % versus 45,5 % après deux ans ; p > 0,05).
Le diagnostic d’épilepsie peut être désormais porté si l’une des conditions suivantes est rencontrée :
- au moins deux crises d’épilepsie non provoquées (ou réflexes) survenues à plus de vingt-quatre heures d’intervalle (définition classique) ;
- une crise d’épilepsie non provoquée (ou réflexe) et une probabilité de récidive d’emblée égale à celle observée après deux crises non provoquées (ou réflexes) [au moins 60 %], et ce sur une période de dix ans à venir après la première crise ;
- un diagnostic d’emblée de syndrome épileptique.
Les crises provoquées restent exclues de la définition de l’épilepsie : ce sont des crises symptomatiques aiguës situationnelles liées à une situation épileptogène transitoire et réversible.4 Le délai requis pour qualifier la crise de « provoquée » après le facteur déclenchant « aigu » est au maximum de sept jours pour les causes toxiques, infectieuses ou lésionnelles (accident cardiovasculaire [AVC], traumatisme crânien, encéphalite ou intervention neurochirurgicale) et de seulement vingt-quatre heures pour les troubles métaboliques et/ou ioniques. Cela signifie concrètement l’abstention thérapeutique et la correction, si cela est possible, du facteur déclenchant. En clair, une crise d’épilepsie survenant sept jours après un AVC ou une encéphalite, ou dans les vingt-quatre heures de la constatation d’une hyponatrémie est considérée comme provoquée et ne conduit pas à l’instauration d’un traitement antiépileptique. Concernant les troubles métaboliques et/ou ioniques, des valeurs seuils ont été fixées par l’ILAE pour retenir l’imputabilité et qualifier ainsi la crise de provoquée : glycémie inférieure à 36 mg/dL (2 mmol) ou supérieure à 450 mg/dL (25 mmol) avec acidocétose, natrémie inférieure à 115 mg/dL, calcémie inférieure à 5 mg/dL ou encore magnésémie inférieure à 0,8 mg/dL.
On peut désormais porter un diagnostic d’épilepsie dès la première crise si l’on estime que le risque de survenue d’une seconde crise est élevé (supérieur à 60 %) et demeure réel sur une période prolongée (dix ans). Une revue Cochrane récente a estimé le risque de récidive après une première crise non provoquée.5 Le premier constat est que la majorité des études des taux et facteurs de récidive portent sur des périodes de deux ans, maximum cinq ans après la première crise, et que l’on dispose de très peu de données sur la récidive à dix ans (trois études seulement). Globalement, sur 46 études incluses dans la méta-analyse, le taux de récurrence des crises était de 27 % à six mois, de 36 % à un an et de 43 % à deux ans, avec des taux légèrement supérieurs de récidive chez l’enfant. D’après une méta-analyse portant sur dix études de classes I et II et sur 3 212 patients, les facteurs de risque établis de récidive, permettant ainsi de porter le diagnostic d’épilepsie dès la première crise, sont : la notion d’une agression cérébrale préexistante (par exemple un antécédent de traumatisme crânien ou d’AVC), des anomalies épileptiques sur l’électroencéphalogramme (EEG) et une imagerie cérébrale anormale.6 Un examen neurologique anormal a également été identifié comme facteur significatif de risque de récidive.
Des conséquences thérapeutiques peuvent découler de cette nouvelle définition élargie, avec des mises en route plus rapides de traitement. Les premiers (et petits) essais contrôlés randomisés de courte durée ont suggéré une réduction apparente de la récurrence des crises dans le cadre d’un traitement immédiat d’une première crise par rapport à un traitement différé. Cet avantage initial disparaît lors d’un suivi plus long avec des taux quasi identiques de rémission à trois ans et à cinq ans chez les patients dont le traitement a été initié ou retardé après une première crise non provoquée, comme l’a démontré l’étude MESS FIRST.7 Une étude récente a analysé l’effet de la définition révisée de l’épilepsie sur les décisions thérapeutiques et les taux de récurrence des crises après une première crise d’épilepsie.8 Il ressort de cette analyse de 629 patients que la proportion d’entre eux recevant un traitement antiépileptique a augmenté de manière significative, passant de 70,4 à 80,5 % (p = 0,015) à la suite de la nouvelle définition de l’épilepsie, sans changement significatif du taux de récidive (40,8 % versus 45,5 % après deux ans ; p > 0,05).
Définition de l’état de mal épileptique
L’état de mal épileptique est défini par la non-cessation d’une crise au bout d’un certain délai ou par la répétition sur ce même délai de deux crises sans reprise de conscience entre les deux. Le délai pour parler d’état de mal et non plus de crise a été fixé par l’ILAE à :9
- cinq minutes pour les crises tonico-cloniques (crises convulsives généralisées d’emblée ou secondairement généralisées) ;
- dix minutes pour les crises focales avec rupture de conscience ;
- entre dix et quinze minutes pour les absences (crises généralisées).
Lire aussi | Anxiété et épilepsie : Likozam disponible en ville
Classification en crises et syndromes
Nouvelle classification des crises depuis 2017
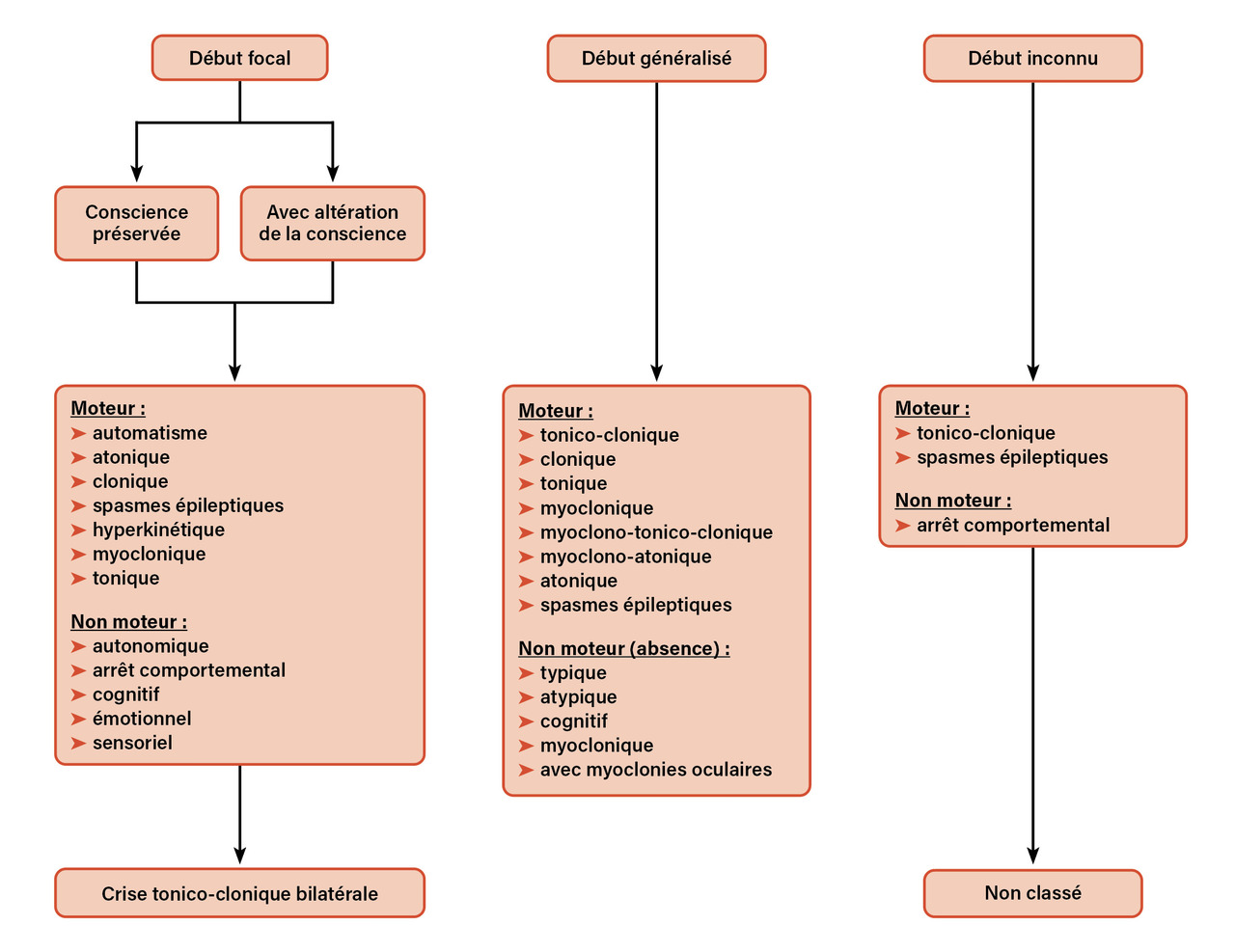
Les crises d’épilepsie sont soit généralisées (vaste réseau cortical ou cortico-sous-cortical bihémisphérique d’emblée), soit focales (départ dans une zone cérébrale donnée avec éventuelle extension secondaire à d’autres régions cérébrales). Dans certains cas, les données cliniques et/ou électroencéphalographiques ne permettent pas de classer les crises (par exemple, crises survenant la nuit avec un EEG normal) ; on parle alors de crise à début inconnu.
L’ILAE a proposé en 2017 une nouvelle classification des crises d’épilepsie (fig. 1 ).10
Les crises focales sont plus fréquentes (deux tiers des crises d’épilepsie). On distingue :
L’ILAE a proposé en 2017 une nouvelle classification des crises d’épilepsie (
Les crises focales sont plus fréquentes (deux tiers des crises d’épilepsie). On distingue :
- les crises focales avec conscience préservée (ex-crises partielles simples) des crises focales avec altération de conscience (ex-crises partielles complexes), potentiellement plus invalidantes et dangereuses ;
- les crises focales avec symptômes moteurs ou non moteurs (principalement des crises avec conscience préservée, sauf celles avec arrêt comportemental). Les symptômes cliniques dépendent de la zone cérébrale concernée par la décharge épileptique ;
- les crises focales évoluant avec une crise tonico-clonique bilatérale ou une « généralisation secondaire ».
- les crises avec symptômes moteurs (tonico-cloniques, myocloniques, cloniques, toniques, atoniques principalement) ;
- les absences.
Classification des syndromes en trois niveaux
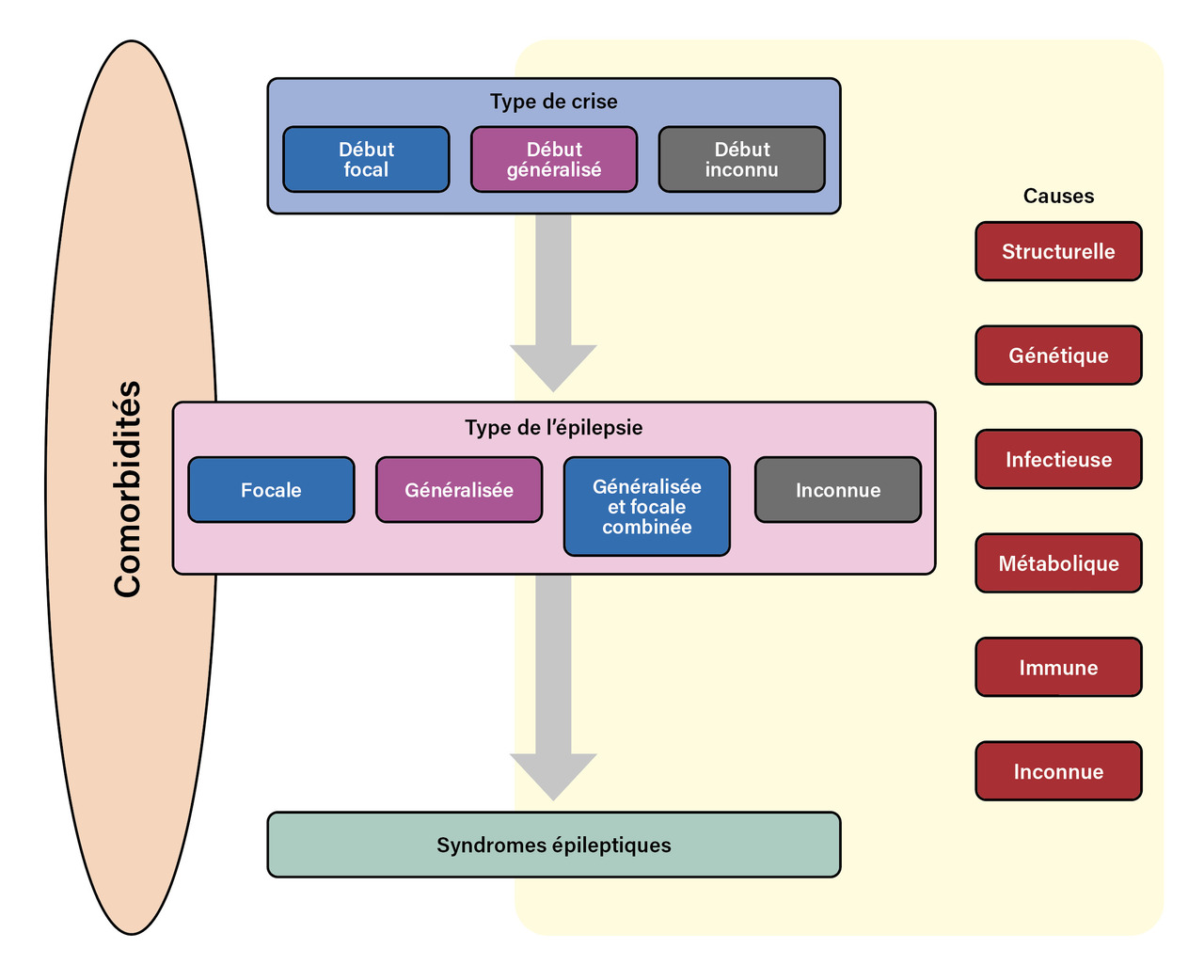
Les syndromes épileptiques, tout comme les crises, sont classés en premier niveau en syndromes généralisés, focaux ou non classés, avec une quatrième catégorie : les syndromes associant des crises généralisées et focales. Historiquement, le second niveau de définition du syndrome épileptique était sa cause générale présumée : idiopathique (présumée polygénique), structurelle-métabolique ou de cause inconnue.
En 2017, l’ILAE a proposé une nouvelle version de la classification avec trois niveaux (fig. 2 ) :11
Dans cette nouvelle classification, les auteurs proposent de ne garder l’appellation « épilepsie généralisée idiopathique » que pour quatre syndromes : épilepsie-absence de l’enfant, épilepsie-absence de l’adolescence, épilepsie myoclonique juvénile, épilepsie avec crises tonico-cloniques exclusives (du réveil).
En 2022, l’ILAE a publié quatre autres articles introduisant une nouvelle terminologie pour certains syndromes.12-14 Cette nouvelle classification est plus axée sur le lien avec l’âge de début (néonatal et nourrisson, enfant, âge variable) et introduit le concept de syndromes spécifiques d’une cause donnée, essentiellement génétique. Les épilepsies généralisées idiopathiques telles que définies en 2017 font l’objet d’une description électroclinique claire et de drapeaux rouges devant faire remettre en cause ou écarter le diagnostic.
En 2017, l’ILAE a proposé une nouvelle version de la classification avec trois niveaux (
- niveau 1 : définition du type de crises ;
- niveau 2 : définition du type d’épilepsie (focale, généralisée, focale et généralisée, inconnue) ;
- niveau 3 : définition du syndrome épileptique.
Dans cette nouvelle classification, les auteurs proposent de ne garder l’appellation « épilepsie généralisée idiopathique » que pour quatre syndromes : épilepsie-absence de l’enfant, épilepsie-absence de l’adolescence, épilepsie myoclonique juvénile, épilepsie avec crises tonico-cloniques exclusives (du réveil).
En 2022, l’ILAE a publié quatre autres articles introduisant une nouvelle terminologie pour certains syndromes.12-14 Cette nouvelle classification est plus axée sur le lien avec l’âge de début (néonatal et nourrisson, enfant, âge variable) et introduit le concept de syndromes spécifiques d’une cause donnée, essentiellement génétique. Les épilepsies généralisées idiopathiques telles que définies en 2017 font l’objet d’une description électroclinique claire et de drapeaux rouges devant faire remettre en cause ou écarter le diagnostic.
Syndromes épileptiques du nouveau-né et du nourrisson
Les syndromes épileptiques du nouveau-né et du nourrisson sont divisés en syndromes autolimités, pour lesquels une rémission spontanée est probable, et en encéphalopathies épileptiques et développementales, pour lesquelles il existe un trouble du développement lié à la fois à la cause sous-jacente et à l’activité épileptique. Il existe une classe émergente de syndromes épileptiques à cause spécifique : celle-ci est associée à un phénotype clinique clairement défini, relativement uniforme et distinct chez la plupart des individus affectés, ainsi qu’à des corrélats – EEG, neuro-imagerie et/ou génétique – cohérents. Le nombre de syndromes définis par leur cause continuera d’augmenter et ces syndromes nouvellement décrits seront à terme incorporés dans cette classification.Syndromes épileptiques débutant dans l’enfance
Les syndromes épileptiques débutant dans l’enfance sont divisés en trois catégories :- les épilepsies focales autolimitées (anciennement épilepsies focales idiopathiques), comprenant quatre syndromes : épilepsie autolimitée avec pointes centro-temporales (SeLECTS, anciennement épilepsie à paroxysmes rolandiques), épilepsie autolimitée avec crises autonomes (SeLEAS, anciennement syndrome de Panayiotopoulos), épilepsie visuelle occipitale infantile (COVE, anciennement épilepsie occipitale de type Gastaut) et épilepsie photosensible du lobe occipital (POLE) ;
- les épilepsies généralisées, comprenant trois syndromes : épilepsie-absence de l’enfant, épilepsie avec absence myoclonique et épilepsie avec myoclonies des paupières ;
- les encéphalopathies développementales et/ou épileptiques, comprenant cinq syndromes : l’épilepsie avec crises myocloniques-atoniques (anciennement syndrome de Doose), le syndrome de Lennox-Gastaut, l’encéphalopathie développementale et/ou épileptique avec activation des pointes-ondes continues du sommeil (anciennement POCS), le syndrome hémiconvulsion-hémiplégie-épilepsie et le syndrome épileptique lié à une infection fébrile (FIRES).
Syndromes épileptiques se manifestant à un âge de début variable
Les syndromes épileptiques se manifestant à un âge variable se répartissent globalement en groupes suivants :- syndromes épileptiques généralisés idiopathiques ;
- syndromes d’épilepsie focale autolimités avec hérédité complexe présumée : épilepsie visuelle occipitale de l’enfant (COVE) et épilepsie photosensible du lobe occipital (POLE) ;
- syndromes épileptiques focaux de cause génétique, structurelle ou génético-structurelle : épilepsie hypermotrice (hyperkinétique) liée au sommeil (SHE), épilepsie familiale du lobe temporal mésial (FMTLE), épilepsie focale familiale à foyers variables (FFEVF) et épilepsie à caractéristiques auditives (EAF) ;
- syndrome d’épilepsie généralisée et focale combiné avec une cause polygénique : épilepsie avec crises induites par la lecture (EwRIS) ;
- syndromes épileptiques avec encéphalopathie de développement (DE), encéphalopathie épileptique (EE) ou les deux, et syndromes épileptiques avec détérioration neurologique progressive : épilepsies myocloniques progressives (PME) et syndrome épileptique lié à une infection fébrile (FIRES) ;
- syndromes épileptiques spécifiques d’une cause : épilepsie du lobe temporal mésial avec sclérose hippocampique et syndrome de Rasmussen.
Lire aussi | Notre dossier – Épilepsie de l’adulte
Incidence plus élevée aux âges extrêmes
L’épilepsie est la maladie neurologique la plus fréquente au monde et la deuxième en France, après les maladies neurodégénératives. Elle affecte, en France, environ 600 000 patients.
Une méta-analyse récente retrouve un taux d’incidence poolée de 61,4 pour 100 000 personnes-années (intervalle de confiance à 95 % [IC 95 %] : 50,7-74,4) avec des disparités :15
Une méta-analyse récente retrouve un taux d’incidence poolée de 61,4 pour 100 000 personnes-années (intervalle de confiance à 95 % [IC 95 %] : 50,7-74,4) avec des disparités :15
- incidence plus élevée aux âges extrêmes de la vie et notamment chez le sujet âgé avec 100 cas pour 100 000 habitants dès 65 ans et une augmentation au fil du temps (taux d’incidence les plus hauts après 80 ans : 180 pour 100 000) ;16
- incidence plus élevée dans les pays à bas ou faible niveau économique comparée aux pays à haut niveau économique : 139 (IC 95 % : 69,4-278,2) vs 48,9 (IC 95 % : 39-61,1) ;
- atteinte des deux sexes, avec une très légère prépondérance masculine.
Références
1. Łukasiuk K, Lasoń W. Emerging molecular targets for anti-epileptogenic and epilepsy modifying drugs. Int J Mol Sci 2023;24(3):2928.
2. Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: A practical clinical definition of epilepsy. Epilepsia 2014;55(4):475-82.
3. Beghi E, Carpio A, Forsgren L, et al. Recommendation for a definition of acute symptomatic seizure. Epilepsia 2010;51(4):671-5.
4. Moshé SL, Perucca E, Ryvlin P, et al. Epilepsy: New advances Lancet 2015;385(9971):884-98.
5. Neligan A, Adan G, Nevitt SJ, et al. Prognosis of adults and children following a first unprovoked seizure. Cochrane Database Syst Rev 2023;1(1):CD013847.
6. Krumholz A, Wiebe S, Gronseth GS, et al. Evidence-based guideline: Management of an unprovoked first seizure in adults: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2015;84(16):1705-13.
7. Kim LG, Johnson TL, Marson AG, et al; MRC MESS Study group. Prediction of risk of seizure recurrence after a single seizure and early epilepsy: Further results from the MESS trial. Lancet Neurol 2006;5(4):317-22.
8. Linka L, Magnus B, Habermehl L, et al. Effect of the revised definition of epilepsy on treatment decisions and seizure recurrence after a first epileptic seizure. Eur J Neurol 2023;30(6):1557-64.
9. Trinka T, Cock H, Hesdorffer D, et al. A definition and classification of status epilepticus. Report of the ILAE Task Force on Classification of Status Epilepticus Epilepsia 2015;56(10):1515-23.
10. Fisher RS, Cross JH, French JA et al. Operational classification of seizure types by the International League Against Epilepsy: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58(4):522-30.
11. Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58(4):512-21.
12. Zuberi SM, Wirrell E, Yozawitz E, et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022;63(6):1349-97.
13. Specchio N, Wirrell EC, Scheffer IE, et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022;63(6):1398-442.
14. Riney K, Bogacz A, Somerville E, et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset at a variable age: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022;63(6):1443-74.
15. Beghi E. The epidemiology of epilepsy. Neuroepidemiology 2020;54(2):185-91.
16. Beghi E, Giussani G, Costa C, et al. The ILAE Task Force on Epilepsy in the Elderly (2017– 2021). The epidemiology of epilepsy in older adults: A narrative review by the ILAE Task Force on Epilepsy in the Elderly. Epilepsia 2023,64:586-601.
2. Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: A practical clinical definition of epilepsy. Epilepsia 2014;55(4):475-82.
3. Beghi E, Carpio A, Forsgren L, et al. Recommendation for a definition of acute symptomatic seizure. Epilepsia 2010;51(4):671-5.
4. Moshé SL, Perucca E, Ryvlin P, et al. Epilepsy: New advances Lancet 2015;385(9971):884-98.
5. Neligan A, Adan G, Nevitt SJ, et al. Prognosis of adults and children following a first unprovoked seizure. Cochrane Database Syst Rev 2023;1(1):CD013847.
6. Krumholz A, Wiebe S, Gronseth GS, et al. Evidence-based guideline: Management of an unprovoked first seizure in adults: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2015;84(16):1705-13.
7. Kim LG, Johnson TL, Marson AG, et al; MRC MESS Study group. Prediction of risk of seizure recurrence after a single seizure and early epilepsy: Further results from the MESS trial. Lancet Neurol 2006;5(4):317-22.
8. Linka L, Magnus B, Habermehl L, et al. Effect of the revised definition of epilepsy on treatment decisions and seizure recurrence after a first epileptic seizure. Eur J Neurol 2023;30(6):1557-64.
9. Trinka T, Cock H, Hesdorffer D, et al. A definition and classification of status epilepticus. Report of the ILAE Task Force on Classification of Status Epilepticus Epilepsia 2015;56(10):1515-23.
10. Fisher RS, Cross JH, French JA et al. Operational classification of seizure types by the International League Against Epilepsy: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58(4):522-30.
11. Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58(4):512-21.
12. Zuberi SM, Wirrell E, Yozawitz E, et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022;63(6):1349-97.
13. Specchio N, Wirrell EC, Scheffer IE, et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022;63(6):1398-442.
14. Riney K, Bogacz A, Somerville E, et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset at a variable age: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022;63(6):1443-74.
15. Beghi E. The epidemiology of epilepsy. Neuroepidemiology 2020;54(2):185-91.
16. Beghi E, Giussani G, Costa C, et al. The ILAE Task Force on Epilepsy in the Elderly (2017– 2021). The epidemiology of epilepsy in older adults: A narrative review by the ILAE Task Force on Epilepsy in the Elderly. Epilepsia 2023,64:586-601.
Dans cet article

Résumé
Les syndromes épileptiques peuvent avoir un âge de début caractéristique du syndrome (nouveau-né, nourrisson, enfant) ou, pour d’autres syndromes, un âge de début variable. On assiste actuellement à l’émergence de syndromes épileptiques spécifiques d’une cause donnée, souvent génétique.