Pourtant, en parallèle des progrès réalisés, il nʼest pas forcément évident, pour le clinicien confronté à un patient souffrant dʼépilepsie inexpliquée, de savoir comment lʼensemble des connaissances actuelles sʼarticule en pratique avec le choix des tests à réaliser, ainsi quʼavec leur utilité, leurs limites et leur interprétation. De fait, les tests génétiques proposés de nos jours pour les patients atteints dʼépilepsie ont évolué et évoluent encore ; ils comprennent différentes techniques cliniquement disponibles. Même si la plupart des épilepsies monogéniques (
Gènes en cause
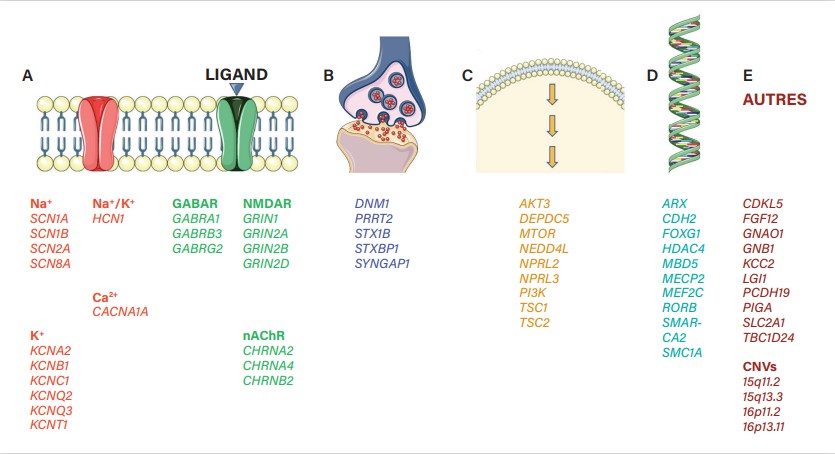
La liste des gènes dont les variations sous-tendent les épilepsies humaines ne cesse de sʼallonger (
- les épilepsies monogéniques causées par une variation dans un gène donné et pouvant suivre des modes de transmission autosomique (dominant ou récessif), liée à lʼX, mitochondriale, ou encore pouvant survenir de novo. Bien que rares, ces épilepsies monogéniques sont les principales cibles du diagnostic génétique ;
- bien plus fréquentes, les épilepsies génétiques complexes, polygéniques, voire multifactorielles (effets de lʼenvironnement), représentent la majorité des épilepsies génétiques généralisées et des épilepsies focales non acquises. Si certains des facteurs de risque génétiques (facteurs de susceptibilité) liés à ces épilepsies complexes commencent à être connus, ils restent encore peu utiles en pratique clinique du fait de la complexité des mécanismes et des interactions en jeu.
Méthodes dʼanalyse génétique
Analyses chromosomiques (caryotype, puces à ADN)
La détection de variations de nombre de copies (CNV) [
Séquençage classique de type Sanger (gène unique)
Séquençage de nouvelle génération (NGS)
Certains centres privilégient un séquençage complet suivi dʼune analyse bio-informatique plus ciblée, focalisée sur un panel défini de gènes-candidats afin de réduire la complexité des interprétations des variants génétiques détectés, tout en gardant ouverte la possibilité de revenir sur les données de NGS plus complètes. Le séquençage du génome, plus complet évidemment que le séquençage dʼexome, génère beaucoup plus de variants dont la signification pathogène est inconnue. Dans les deux modalités (exome et génome), le séquençage des parents en plus de celui du patient (séquençages de trios) facilite grandement les interprétations.
Cas particuliers
Indications et choix des méthodes
Quels types dʼépilepsie et quels patients tester ? Quʼen est-il des adultes ?
En pratique clinique, les tests génétiques les plus utiles concernent les épilepsies les plus sévères, pharmacorésistantes. Lʼindication qui semble la plus évidente est celle des DEE et des épilepsies survenant dans un contexte neurodéveloppemental (comorbidité avec des troubles du spectre autisti-que, avec un retard intellectuel...) ; un contexte dysmorphique ou une atteinte multisystémique rend aussi le test génétique particulièrement pertinent. Dans les formes familiales dʼépilepsies de lʼenfance autolimitées,7 le test génétique peut faciliter le diagnostic et préciser le pronostic et le risque de récurrence, tout en prenant en compte la possibilité de pénétrance incomplète et dʼexpressivité clinique variable. Dans le cas des épilepsies focales ou généralisées sporadiques, voire isolées, le rendement du test génétique reste généralement faible.
En gardant à lʼesprit que le jugement du clinicien doit sʼadapter à la particularité de chaque cas et apprécier les bénéfices et limites du test génétique selon la situation, quelques points méritent dʼêtre soulignés : ainsi, les nouveau-nés et les nourrissons présentant des crises et les cas déjà mentionnés plus haut avec DEE ou épilepsie dans un contexte de pathologie neurodéveloppementale présentent le plus fort rendement de diagnostic génétique. Ce rendement reste cependant toujours appréciable pour les autres tranches dʼâge ; de fait, un âge de début des crises plus tardif ou lʼabsence de comorbidité, par exemple, ne doit pas sʼopposer à lʼindication du test génétique. On peut même considérer que tous les individus avec épilepsie, quel quʼen soit le type, pourraient à terme tirer un bénéfice de la réalisation dʼun test génétique, quʼil sʼagisse dʼadapter un traitement, dʼaffiner le pronostic ou de proposer un conseil génétique.6
De fait, si la probabilité de succès du test diminue avec un âge de début des symptômes plus tardif, lʼâge du patient lui-même au moment où le test est envisagé ne doit pas influencer la décision : un adulte ayant eu une épilepsie à début précoce nʼa certainement pas bénéficié à lʼépoque des connaissances actuelles et des tests disponibles de nos jours. De plus, lʼexistence dʼantécédents périnataux ne doit pas faire exclure une cause génétique, y compris chez lʼadulte, et le diagnostic génétique reste donc indiqué.
Enfin, il faut souligner des études relativement récentes8,9 indiquant que les épileptiques adultes, et plus particulièrement la minorité dʼentre eux qui présente un certain degré de déficience intellectuelle, doivent certainement se voir proposer un test génétique : celui-ci a une probabilité assez élevée de succès (et vouée à augmenter avec lʼamélioration des techniques et lʼaccroissement des connaissances) et peut conduire à des investigations cliniques complémentaires ou à des optimisations thérapeutiques pour certains des patients (syndrome de Dravet ou déficience en GLUT-1, par exemple).
Quels tests utiliser ?
Interprétation des résultats
On distingue schématiquement trois cas de figure :
- la pathologie sʼexplique parfaitement par lʼanomalie génétique détectée et suspectée ;
- le lien de causalité dʼune ou de plusieurs anomalie(s) détectée(s) avec la pathologie reste incertain. Une appréciation plus fine du phénotype, une extension du test aux parents, voire à dʼautres membres de la famille, et des analyses biologiques pour apprécier lʼimpact fonctionnel des anomalies génétiques doivent être envisagées ;
- aucune anomalie génétique détectée ne semble compatible avec une pathogénicité en rapport avec lʼépilepsie. Une réévaluation régulière des résultats avec le temps et des tests génétiques complémentaires peuvent permettre de résoudre le problème, en attendant lʼémergence de nouvelles modalités de tests génétiques.
Intérêt pour le diagnostic et le pronostic avec quelques réserves
Quant aux inconvénients, il peut sʼagir de lʼimpact psychologique sur le patient ou son entourage, du risque de stigmatisation, voire de discrimination (assurances, employabilité). Le risque de découverte incidente, sans rapport initial avec lʼindication du test, que ce soit avec (prévention, surveillance) ou sans possibilité dʼintervenir sur un risque de développer une pathologie donnée, est évidemment à considérer – problème dʼautant plus délicat que les progrès de la médecine peuvent dans le futur modifier les options.
Perspectives
Quelques définitions
Encéphalopathies épileptiques et développementales (developmental and epileptic encephalopathies [DEE])
Épilepsies sévères se manifestant tôt dans la vie, accompagnées dʼun développement psychomoteur anormal dû à la pathologie sous-jacente ainsi quʼà lʼactivité épileptique, dont les contributions relatives peuvent être difficiles à déterminer.
Épilepsies monogéniques
Pour ces épilepsies, une variation dans un seul gène est suffisante pour causer la pathologie chez un patient donné. Un type dʼépilepsie monogénique (par exemple, les épilepsies hypermotrices liées au sommeil) peut être causé, chez des patients différents, par des variants dans des gènes différents (pour lʼexemple cité ici, les gènes CHRNA4, CHRNB2, KCNT1, DEPDC5...).
Hybridation génomique comparative (comparative genomic hybridization [CGH])
Technique de cytogénétique moléculaire sur puce à ADN permettant une analyse globale des variations de nombre de copies de séquences dʼADN dʼun génome donné, par rapport à un génome contrôle.
Séquençages de nouvelle génération (next-generation sequencing [NGS])
Aussi appelées séquençages à haut débit, ces techniques reposent sur lʼanalyse rapide et simultanée dʼun très grand nombre de segments dʼADN.
Variants génétiques de novo
Variants de la lignée germinale présents dans toutes les cellules de lʼindividu et non détectables chez ses parents, survenant généralement durant la gamétogenèse.
Variations de nombre de copies (copy number variations [CNV])
Variations (gain ou perte) de copies dʼune région génomique dont la taille est supérieure à 1 kilobase et le nombre de copies diffère par rapport au génome de référence.
Variants génétiques mosaïques post-zygotiques
Variations génétiques qui ne sont présentes que dans un sous-ensemble (plus ou moins important) des cellules de lʼorganisme.
2. Szepetowski P. Genetics of human epilepsies: Continuing progress. Presse Med 2018;47(3):218-26.
3. Niestroj LM, Perez-Palma E, Howrigan DP, et al. Epilepsy subtype-specific copy number burden observed in a genome-wide study of 17 458 subjects. Brain 2020;143(7):2106-18.
4. Krey I, Platzer K, Esterhuizen A, et al. Current practice in diagnostic genetic testing of the epilepsies. Epileptic Disord 2022;24(5):765-86.
5. Pickrell WO, Fry AE. Epilepsy genetics: A practical guide for adult neurologists. Pract Neurol 2023;23(2):111-9.
6. Smith L, Malinowski J, Ceulemans S, et al. Genetic testing and counseling for the unexplained epilepsies: An evidence-based practice guideline of the National Society of Genetic Counselors. J Genet Couns 2023;32(2):266-80.
7. Specchio N, Wirrell EC, Scheffer IE, et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE task force on nosology and definitions. Epilepsia 2022;63(6):1398-442.
8. Borlot F, de Almeida BI, Combe SL, et al. Clinical utility of multigene panel testing in adults with epilepsy and intellectual disability. Epilepsia 2019;60(8):1661-9.
9. Johannesen KM, Nikanorova N, Marjanovic D, et al. Utility of genetic testing for therapeutic decision-making in adults with epilepsy. Epilepsia 2020;61(6):1234-9.
10. Arnaud L, Abi Warde MT, Barcia G, et al. The EPIGENE network: A French initiative to harmonize and improve the nationwide diagnosis of monogenic epilepsies. Eur J Med Genet 2022;65(3):104445.
11. DʼGama AM, Poduri A. Brain somatic mosaicism in epilepsy: Bringing results back to the clinic. Neurobiol Dis 2023;181:106104.
12. Ellis CA, Petrovski S, Berkovic SF. Epilepsy genetics: Clinical impacts and biological insights. Lancet Neurol 2020;19(1):93-100.
13. Sheidley BR, Malinowski J, Bergner AL, et al. Genetic testing for the epilepsies: A systematic review. Epilepsia 2022;63(2):375-87.
 Encadrés
Encadrés