Une patiente de 86 ans est adressée en rhumatologie pour deux raisons : le bilan de fractures multiples et une lésion lytique du tiers inférieur de l’avant-bras gauche.
Parmi ses antécédents figurent une hypertension artérielle sous traitement, des troubles cognitifs, un méningiome frontal n’ayant pas nécessité d’intervention mais un traitement par corticothérapie. Le contrôle de l’imagerie par résonance magnétique (IRM) cérébrale, un an après le diagnostic, a montré une stabilité de la taille du méningiome. Dans la même année, la patiente a souffert d’une fracture traumatique du col fémoral droit, ayant nécessité la mise en place d’une prothèse totale. Parallèlement, un complément de bilan osseux a fortuitement mis en évidence une fracture du radius droit, traitée orthopédiquement, associée à des fractures vertébrales T11, T12 et L1 d’allure bénigne.
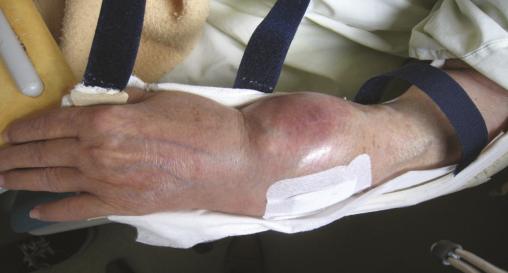
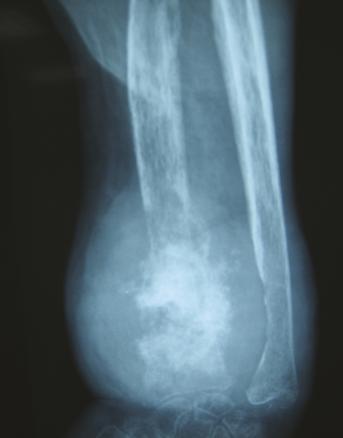
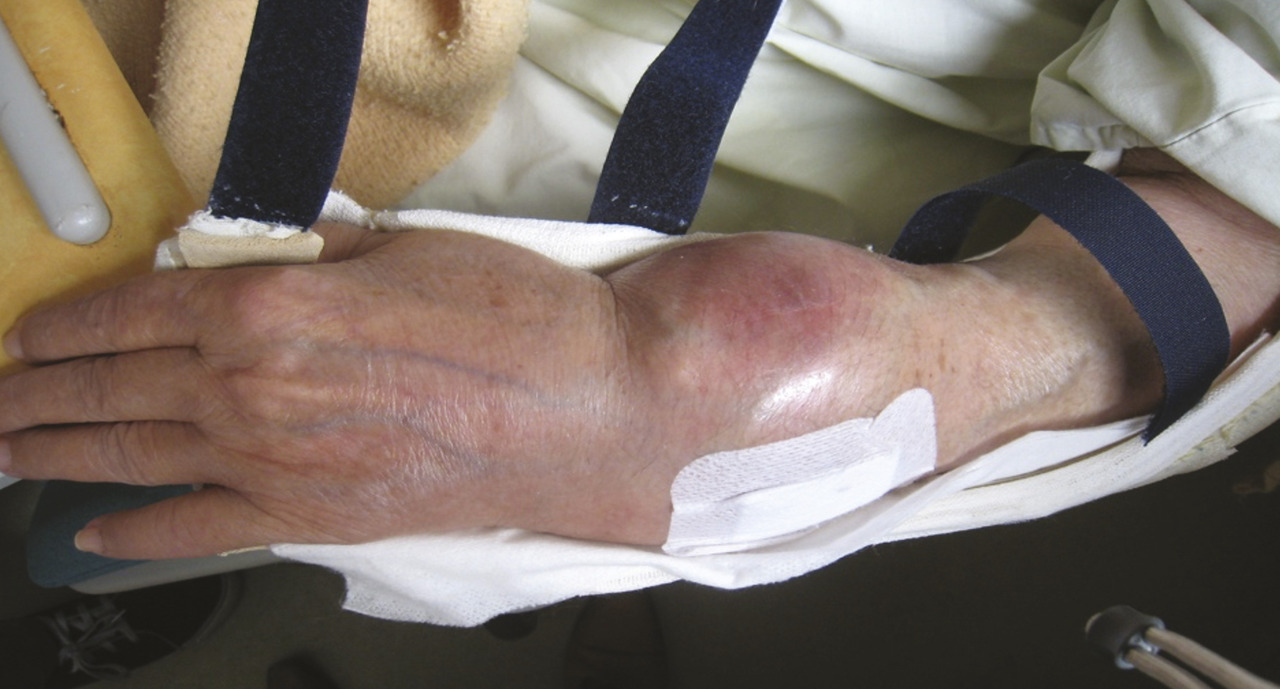
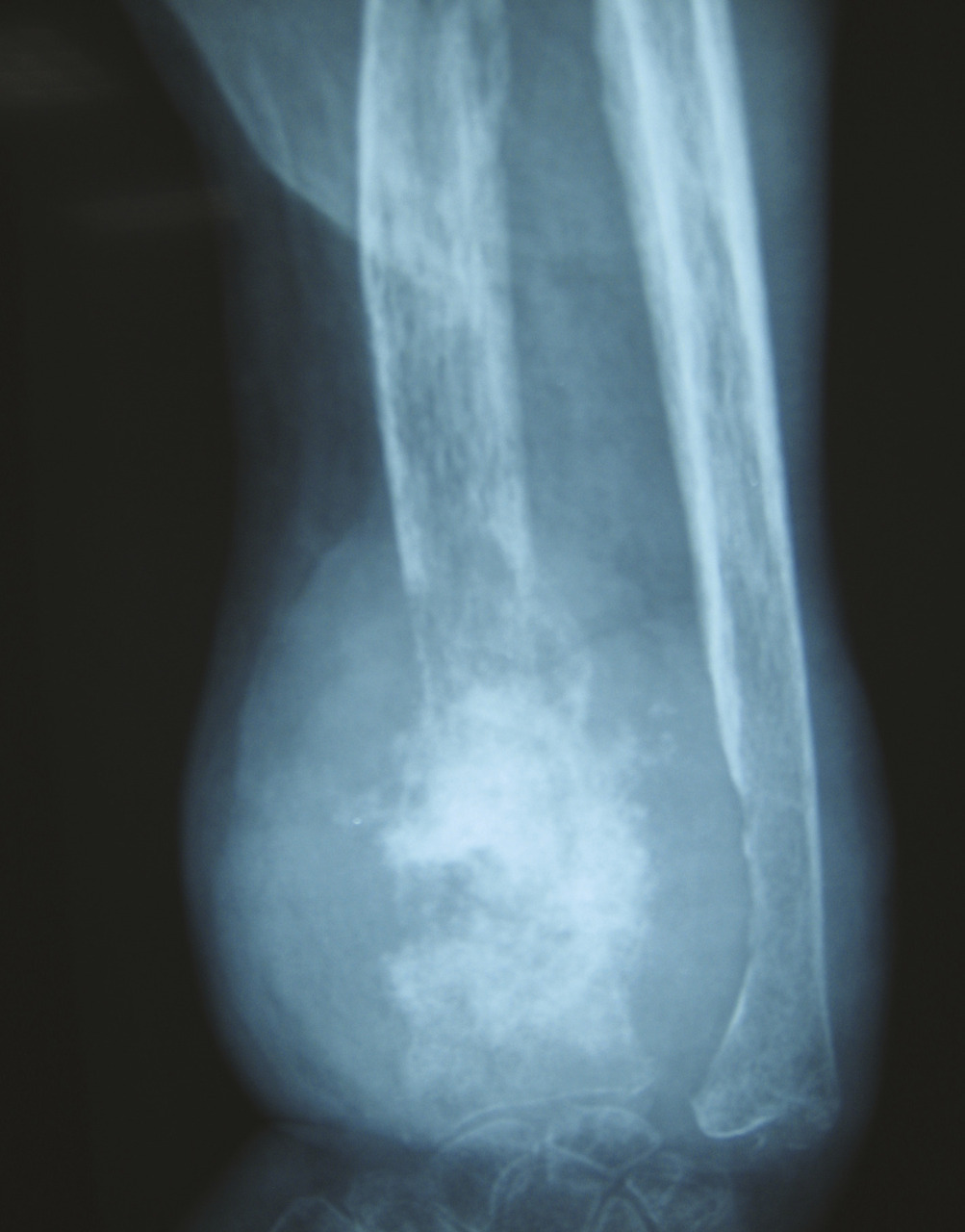
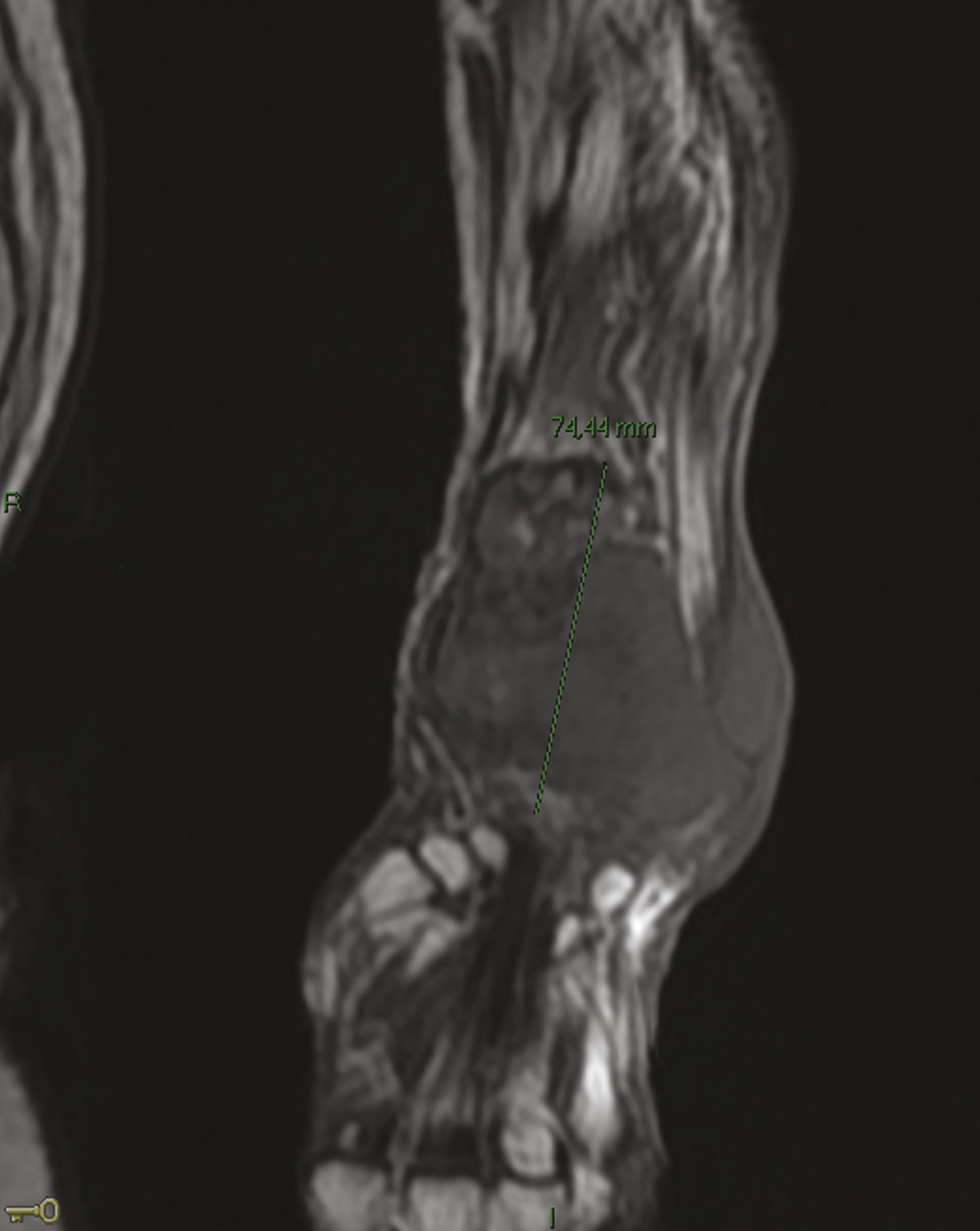
Elle est admise à l’hôpital deux ans plus tard pour des douleurs mixtes et une tuméfaction douloureuse du tiers distal de l’avant-bras gauche, dure et fixe (fig. 1 ). À l’examen physique, la patiente est grabataire et souffre de douleurs osseuses provoquées diffuses, avec une sarcopénie. La radiographie standard met en évidence une importante ostéolyse radiale inférieure, avec rupture des corticales, associée à des images d’ossifications en regard s’étendant aux parties molles épaissies (fig. 2 ). Le même aspect est confirmé sur l’IRM (fig. 3 ). La tomodensitométrie (TDM) thoraco-abdomino-pelvienne est normale. La mammographie ne retrouve pas d’anomalie. La biopsie chirurgicale montre une lésion inhabituelle associant une substance amorphe, une ostéogenèse et des cellules atypiques. Un diagnostic formel n’a pas pu être posé et il n’y avait pas d’arguments pour une origine maligne ou une tumeur osseuse primitive sur le prélèvement. Par ailleurs, le bilan de fractures multiples n’a pas mis en évidence de syndrome inflammatoire biologique. En revanche, une hypophosphorémie a motivé la réalisation d’une scintigraphie osseuse montrant un aspect évocateur d’ostéomalacie.
La patiente a reçu une supplémentation phosphocalcique et de l’alfacalcidol (dérivé de la vitamine D). Un nouveau bilan a montré un taux de phosphore très bas à 0,30 mmol/L. Cette hypophosphorémie normocalcémique était associée à un dosage d’hormone parathyroïdienne (PTH) normal et à une phosphaturie augmentée. Le dosage de la 25-OH vitamine D était normal. L’ensemble de ces éléments plaide en faveur d’un diabète phosphoré sélectif. Le dosage du fibroblast growth factor 23 (FGF23) était trop élevé pour être quantifié. La scintigraphie à l’octréotide a retrouvé une hyperfixation frontale gauche. Devant un tableau de malaise, vomissements, confusion et céphalées, une nouvelle IRM cérébrale a montré un aspect de méningiome avec un discret œdème cérébral, connu depuis 2010.
Le diagnostic retenu est une ostéomalacie oncogénique secondaire à une tumeur sécrétant du FGF23 d’origine cérébrale.
La seule alternative thérapeutique est l’exérèse de la tumeur afin de normaliser la phosphorémie et d’envisager une récupération de l’autonomie. La patiente a été adressée en neurochirurgie pour discuter une intervention ou un traitement médical compte tenu du risque hémorragique lors de l’exérèse du méningiome.
Parmi ses antécédents figurent une hypertension artérielle sous traitement, des troubles cognitifs, un méningiome frontal n’ayant pas nécessité d’intervention mais un traitement par corticothérapie. Le contrôle de l’imagerie par résonance magnétique (IRM) cérébrale, un an après le diagnostic, a montré une stabilité de la taille du méningiome. Dans la même année, la patiente a souffert d’une fracture traumatique du col fémoral droit, ayant nécessité la mise en place d’une prothèse totale. Parallèlement, un complément de bilan osseux a fortuitement mis en évidence une fracture du radius droit, traitée orthopédiquement, associée à des fractures vertébrales T11, T12 et L1 d’allure bénigne.
Elle est admise à l’hôpital deux ans plus tard pour des douleurs mixtes et une tuméfaction douloureuse du tiers distal de l’avant-bras gauche, dure et fixe (
La patiente a reçu une supplémentation phosphocalcique et de l’alfacalcidol (dérivé de la vitamine D). Un nouveau bilan a montré un taux de phosphore très bas à 0,30 mmol/L. Cette hypophosphorémie normocalcémique était associée à un dosage d’hormone parathyroïdienne (PTH) normal et à une phosphaturie augmentée. Le dosage de la 25-OH vitamine D était normal. L’ensemble de ces éléments plaide en faveur d’un diabète phosphoré sélectif. Le dosage du fibroblast growth factor 23 (FGF23) était trop élevé pour être quantifié. La scintigraphie à l’octréotide a retrouvé une hyperfixation frontale gauche. Devant un tableau de malaise, vomissements, confusion et céphalées, une nouvelle IRM cérébrale a montré un aspect de méningiome avec un discret œdème cérébral, connu depuis 2010.
Le diagnostic retenu est une ostéomalacie oncogénique secondaire à une tumeur sécrétant du FGF23 d’origine cérébrale.
La seule alternative thérapeutique est l’exérèse de la tumeur afin de normaliser la phosphorémie et d’envisager une récupération de l’autonomie. La patiente a été adressée en neurochirurgie pour discuter une intervention ou un traitement médical compte tenu du risque hémorragique lors de l’exérèse du méningiome.
L’ostéomalacie oncogénique, rare, fait partie des syndromes paranéoplasiques. Elle se caractérise par une hypophosphorémie sévère induite par des facteurs phosphaturiants, les phosphatonines, principalement le FGF23 sécrété par certaines tumeurs mésenchymateuses.1 Son dosage apporte une aide décisive au diagnostic.
La survenue de fractures sur os ostéomalacique liées à la présence d’une tumeur mésenchymateuse développée à partir des méninges, comme dans le cas de cette patiente, est extrêmement rare. Bien qu’histologiquement hétérogène, la tumeur mésenchymateuse est une variante mixte du tissu conjonctif, développée au niveau de l’os ou les tissus mous. Trois cas de siège intracrânien sont décrits dans la littérature.2,3 L’ostéomalacie est caractérisée par un ramollissement osseux progressif, dû à une minéralisation insuffisante du tissu ostéoïde. Les causes les plus fréquentes comprennent une carence en vitamine D et une dysfonction tubulaire rénale. Cependant, dans de rares cas, l’ostéomalacie peut être associée à des tumeurs des tissus mous (ostéomalacie oncogénique), équivalent d’un syndrome paranéoplasique.4,5 Le mécanisme à l’origine de l’insuffisance de minéralisation du tissu ostéoïde n’est pas entièrement compris, mais la surexpression tumorale de FGF23 y joue un rôle. Cette protéine inhibe le transport du phosphate transépithélial dans les tubules rénaux et entraîne une perte rénale de phosphate et une ostéomalacie ultérieure.6 Le diagnostic est difficilement porté par les examens d’imagerie (TDM et IRM), en raison de la faible taille de la tumeur. La durée moyenne avant le diagnostic est généralement de six ans. L’histologie de la pièce d’exérèse retrouve des zones de calcifications, des ostéoclastes like cellules et un réseau capillaire bien développé. L’immunomarquage auxiliaire avec des anticorps anti-FGF23 et de la dentine protéine de matrice 1 (DMP1) sont également utiles pour confirmer le diagnostic.7
Les patients ont en général une longue histoire de douleurs osseuses et des fractures multiples. Les examens biologiques révèlent une hypophosphatémie, une hyperphosphaturie, l’augmentation de l’activité phosphatase alcaline et une valeur basse ou inappropriée du 1,25- dihydroxy D3, avec une faible réponse à la supplémentation par la vitamine D.8 Bien que rares, les variantes non phosphaturiantes peuvent aussi exister.
L’exérèse chirurgicale complète de la tumeur est curative ; elle entraîne une correction rapide des anomalies métaboliques et la résolution éventuelle de symptômes associés à l’ostéomalacie.8
La survenue de fractures sur os ostéomalacique liées à la présence d’une tumeur mésenchymateuse développée à partir des méninges, comme dans le cas de cette patiente, est extrêmement rare. Bien qu’histologiquement hétérogène, la tumeur mésenchymateuse est une variante mixte du tissu conjonctif, développée au niveau de l’os ou les tissus mous. Trois cas de siège intracrânien sont décrits dans la littérature.2,3 L’ostéomalacie est caractérisée par un ramollissement osseux progressif, dû à une minéralisation insuffisante du tissu ostéoïde. Les causes les plus fréquentes comprennent une carence en vitamine D et une dysfonction tubulaire rénale. Cependant, dans de rares cas, l’ostéomalacie peut être associée à des tumeurs des tissus mous (ostéomalacie oncogénique), équivalent d’un syndrome paranéoplasique.4,5 Le mécanisme à l’origine de l’insuffisance de minéralisation du tissu ostéoïde n’est pas entièrement compris, mais la surexpression tumorale de FGF23 y joue un rôle. Cette protéine inhibe le transport du phosphate transépithélial dans les tubules rénaux et entraîne une perte rénale de phosphate et une ostéomalacie ultérieure.6 Le diagnostic est difficilement porté par les examens d’imagerie (TDM et IRM), en raison de la faible taille de la tumeur. La durée moyenne avant le diagnostic est généralement de six ans. L’histologie de la pièce d’exérèse retrouve des zones de calcifications, des ostéoclastes like cellules et un réseau capillaire bien développé. L’immunomarquage auxiliaire avec des anticorps anti-FGF23 et de la dentine protéine de matrice 1 (DMP1) sont également utiles pour confirmer le diagnostic.7
Les patients ont en général une longue histoire de douleurs osseuses et des fractures multiples. Les examens biologiques révèlent une hypophosphatémie, une hyperphosphaturie, l’augmentation de l’activité phosphatase alcaline et une valeur basse ou inappropriée du 1,25- dihydroxy D3, avec une faible réponse à la supplémentation par la vitamine D.8 Bien que rares, les variantes non phosphaturiantes peuvent aussi exister.
L’exérèse chirurgicale complète de la tumeur est curative ; elle entraîne une correction rapide des anomalies métaboliques et la résolution éventuelle de symptômes associés à l’ostéomalacie.8

Références
1. Fery-Blanco C, Prié D, Gil H, et al. Une ostéomalacie grave miraculeusement guérie par des soins dentaires, cas cliniques et thérapeutiques. Médecine thérapeutique 2005;11(2):342-5.
2. Reis-Filho JS, Paiva ME, Lopes JM. Phosphaturic mesenchymal tumor (oncogenic osteomalacia). Brain Pathol 2004;14(1):111-2, 115.
3. Bower RS, Daugherty WP, Giannini C, et al. Intracranial phosphaturic mesenchymal tumor, mixed connective tissue variant presenting without oncogenic osteomalacia. Surg Neurol Int 2012;3:151.
4. Folpe AL, Fanburg-Smith JC, Billings SD, et al. Most osteomalacia-associated mesenchymal tumors are a single histopathologic entity: An analysis of 32 cases and a comprehensive review of the literature. American Journal of Surgical Pathology 2004;28(1):1-30.
5. Policarpio-Nicolas ML, Abbott TE, Dalkin AC, et al. Phosphaturic mesenchymal tumor diagnosed by fine-needle aspiration and core biopsy: A case report and review of literature. Diagn Cytopathol 2008;36(2):115-9.
6. Yoshioka K, Nagata R, Ueda M, et al. Phosphaturic mesenchymal tumor with symptoms related to osteomalacia that appeared one year after tumorectomy. Intern Med 2006;45(20):1157-60.
7. Toyosawa S, Tomita Y, Kishino M, et al. Expression of dentin matrix protein 1 in tumors causing oncogenic osteomalacia. Mod Pathol 2004;17(5):573-8.
8. Sundaram M, McCarthy EF. Oncogenic osteomalacia. Skeletal Radiol 2000;29(3):117-24.
2. Reis-Filho JS, Paiva ME, Lopes JM. Phosphaturic mesenchymal tumor (oncogenic osteomalacia). Brain Pathol 2004;14(1):111-2, 115.
3. Bower RS, Daugherty WP, Giannini C, et al. Intracranial phosphaturic mesenchymal tumor, mixed connective tissue variant presenting without oncogenic osteomalacia. Surg Neurol Int 2012;3:151.
4. Folpe AL, Fanburg-Smith JC, Billings SD, et al. Most osteomalacia-associated mesenchymal tumors are a single histopathologic entity: An analysis of 32 cases and a comprehensive review of the literature. American Journal of Surgical Pathology 2004;28(1):1-30.
5. Policarpio-Nicolas ML, Abbott TE, Dalkin AC, et al. Phosphaturic mesenchymal tumor diagnosed by fine-needle aspiration and core biopsy: A case report and review of literature. Diagn Cytopathol 2008;36(2):115-9.
6. Yoshioka K, Nagata R, Ueda M, et al. Phosphaturic mesenchymal tumor with symptoms related to osteomalacia that appeared one year after tumorectomy. Intern Med 2006;45(20):1157-60.
7. Toyosawa S, Tomita Y, Kishino M, et al. Expression of dentin matrix protein 1 in tumors causing oncogenic osteomalacia. Mod Pathol 2004;17(5):573-8.
8. Sundaram M, McCarthy EF. Oncogenic osteomalacia. Skeletal Radiol 2000;29(3):117-24.
Une question, un commentaire ?