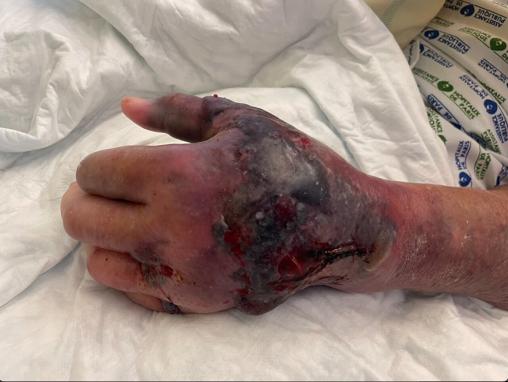
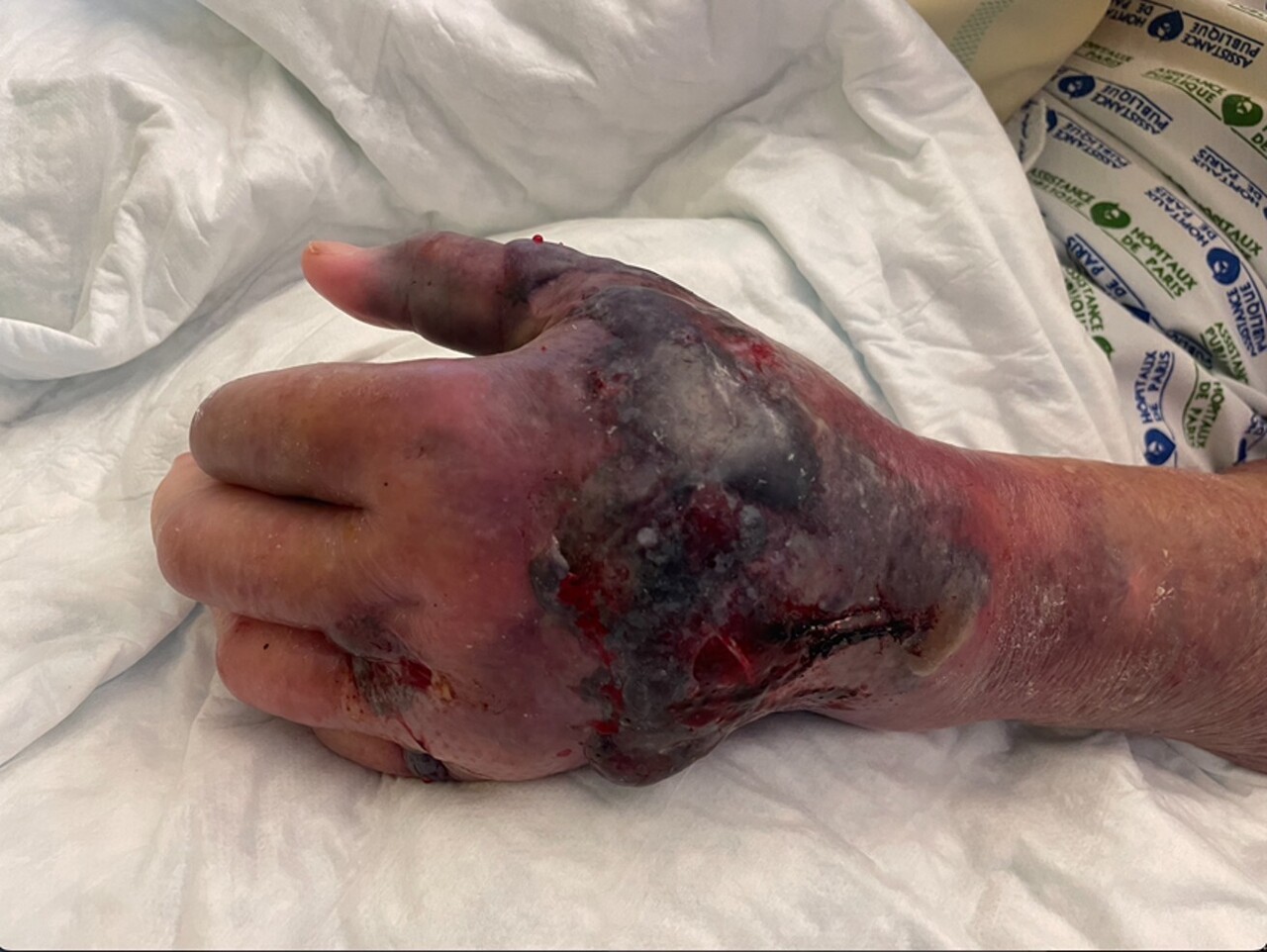
L’examen clinique révèle des ecchymoses diffuses au niveau du thorax et des membres inférieurs et un important hématome de la main gauche d’allure plus récente (figure).
La radiographie du membre ne montre aucune fracture. Il existe une anémie normocytaire arégénérative à 10,2 g/dL, le taux de prothrombine est à 88 %, le temps de céphaline activée (TCA) est allongé à 3,04.
Le facteur VIII est effondré, sans anomalies des autres facteurs de la coagulation. Le mélange du plasma de la patiente avec un plasma de témoin confirme la présence d’un anticorps circulant. Le dosage spécifique de l’anticorps anti-facteur VIII est positif à taux élevé (357 BU/mL).
L’hémophilie acquise A est une maladie rare mais constitue le plus fréquent des troubles acquis de la coagulation. Elle concerne principalement les sujets âgés. Elle s’explique par la production d’anticorps dirigés contre le facteur VIII de la coagulation et peut s’associer à une maladie auto-immune ou à une néoplasie.1
Elle est responsable de saignements spontanés ou provoqués par un traumatisme minime.
Son diagnostic repose sur l’exploration du TCA allongé : confirmer l’allongement (temps de céphaline activée allongé), éliminer l’utilisation d’héparine (temps de thrombine normal), confirmer un déficit quantitatif en facteur de la coagulation, puis préciser le mécanisme auto-immun par le mélange avec plasma de témoin, où le TCA n’est pas corrigé, signant la présence d’un anticorps — et non d’un simple déficit en facteur de la coagulation. Enfin, le dosage de l’anticorps antifacteur VIII par ELISA et sa titration permettent de confirmer le diagnostic et de suivre la maladie.
La prise en charge thérapeutique repose sur l’injection de facteur VII activé afin de contourner la voie intrinsèque défaillante ; le traitement étiologique associe une corticothérapie au rituximab (anticorps anti-CD20)2 ou au cyclophosphamide. Un nouvel anticorps bispécifique dirigé contre les facteurs IX et X, l’émicizumab, permet de suppléer le rôle du facteur VIII en attendant l’efficacité des immunosuppresseurs sans être lui-même ciblé par les anticorps du patient.3,4
L’utilisation d’un tel protocole a permis à la patiente un tarissement des saignements après quarante-huit heures, avec une normalisation progressive du TCA.
2. Tiede A, Collins P, Knoebl P, et al. International recommendations on the diagnosis and treatment of acquired hemophilia A. Haematologica 2020;105(7):1791‑801.
3. Mahlangu J, Oldenburg J, Paz-Priel I, et al. Emicizumab prophylaxis in patients who have hemophilia a without inhibitors. N Engl J Med 2018;379(9):811‑22.
4. Denis CV, Lenting PJ. Functional and clinical aspects of the anti-hemophilic bispecific antibody emicizumab. Hématologie 2020;26(6):328‑42.
Une question, un commentaire ?
