Un consensus a été rédigé en 2024 sous l’égide de la Société française d’endocrinologie, de l’Association francophone de chirurgie endocrinienne et de la Société française de médecine nucléaire, relatif aux diagnostics positif, étiologique, différentiel et au traitement de l’hyperparathyroïdie primaire (HPT1). Une synthèse à destination des spécialistes en médecine générale est ici proposée.
Physiologiquement, la parathormone (ou hormone parathyroïdienne, PTH) régule l’homéostasie calcique en stimulant l’absorption intestinale du calcium, la résorption osseuse et la réabsorption rénale du calcium, via le récepteur membranaire sensible au calcium (CaSR). Par l’intermédiaire de ce récepteur présent sur les cellules parathyroïdiennes et le tubule rénal, l’augmentation de la calcémie ionisée inhibe la sécrétion de la PTH et la réabsorption rénale du calcium, induisant une hypercalciurie.
HPT1 en quelques chiffres
L’HPT1 est une pathologie fréquente, avec une prévalence évaluée entre 17 et 94,6 pour 100 000 patients, notamment chez la femme après la ménopause où elle atteint 1,5 %. Son incidence semble stabilisée dans les pays occidentaux (calcémie en routine, bilan étiologique d’ostéoporose) tandis que sa prévalence s’accroît du fait de l’augmentation de l’espérance de vie.1
Signes et contexte
Le diagnostic d’HPT1 est biologique ; les symptômes sont devenus rarement révélateurs du fait d’un dosage plus systématisé de la calcémie.
Diagnostic positif
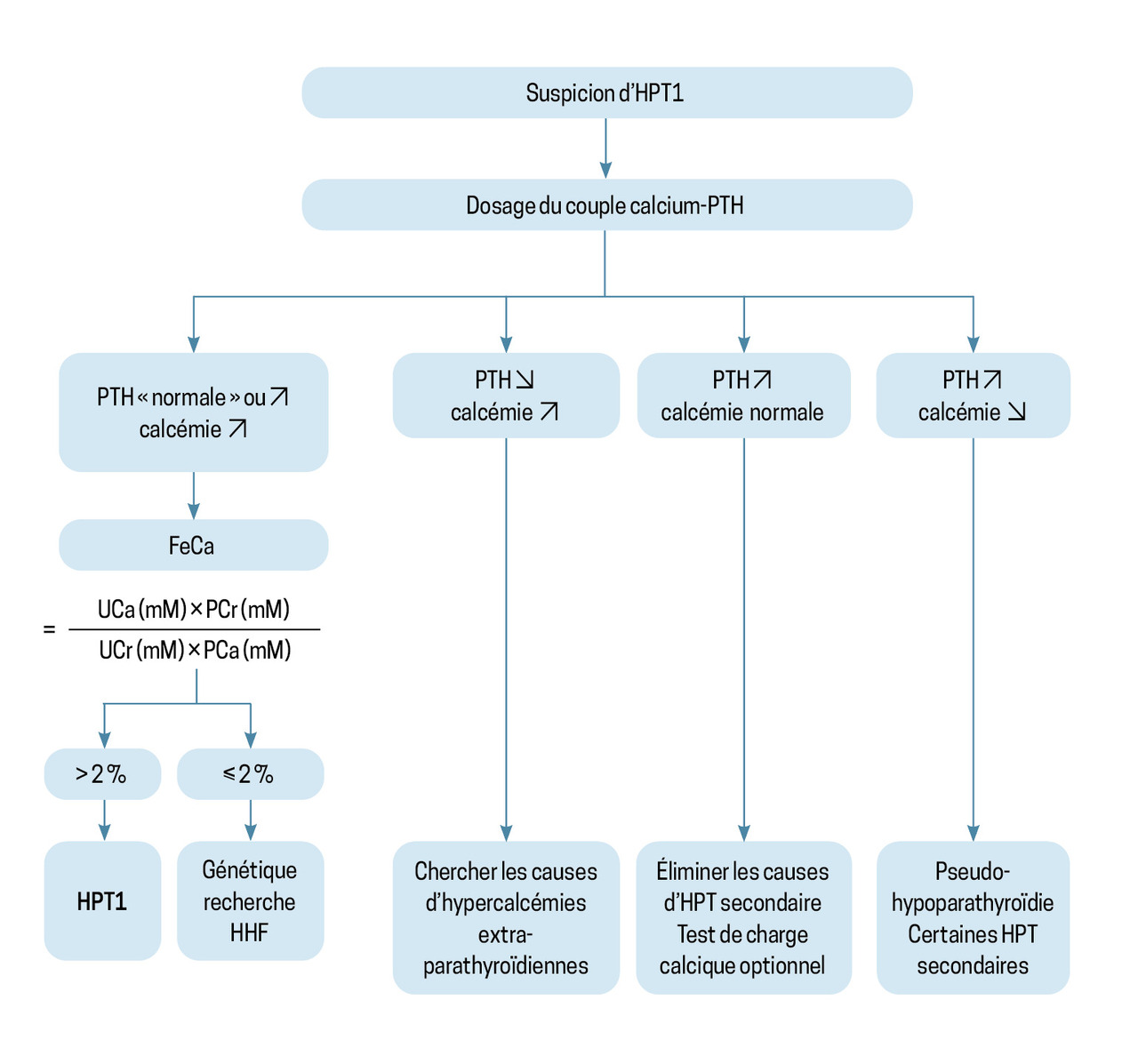
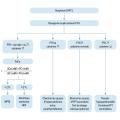
Le diagnostic positif d’HPT1 repose sur une valeur de PTH trop élevée par rapport à la valeur de la calcémie.2 La forme typique associe une hypercalcémie, une ascension de la PTH et une hypercalciurie. Les formes atypiques associent soit une hypercalcémie et une parathormonémie normale, soit une calcémie normale avec une hyperparathormonémie, qui ne peut pas être secondaire à une autre cause (comme une insuffisance vitaminique D). Les formes atypiques nécessitent l’avis d’un spécialiste en endocrinologie (fig. 1).
Retentissement clinique de l’hyperparathyroïdie
L’HPT1 est devenue une pathologie majoritairement asymptomatique grâce à la mesure plus systématisée de la calcémie. Son diagnostic formel impose donc de rechercher une atteinte des organes cibles, lorsque cette dernière n’est pas révélatrice de l’HPT1.
Il existe des signes classiques et des signes non classiques d’HPT1.2
Les signes classiques correspondent aux signes osseux manifestes (douleur osseuse, tumeur brune, kystes), rénaux (lithiase rénale, néphrocalcinose) et musculaires (myopathie proximale) [tableau 1]. Il existe un lien de causalité entre ces signes et l’HPT1, et ils sont réversibles après chirurgie.3
Les signes non classiques (cardiovasculaires à type d’hypertension artérielle, digestifs [anorexie, constipation], neuropsychiques), surtout mis en évidence dans des études d’association,2 ne régressent pas nécessairement après parathyroïdectomie et peuvent survenir pour des hypercalcémies mineures.
Leur présence impose néanmoins de réaliser une mesure de la calcémie.
Enquête étiologique
Une fois le diagnostic biologique affirmé ou fortement suspecté, l’enquête étiologique implique d’une part une démarche de diagnostic différentiel, d’autre part une réflexion sur la cause sporadique ou potentiellement génétique, souvent marquée par une atteinte multiglandulaire.
Diagnostic différentiel
Les diagnostics différentiels de l’HPT1 peuvent s’envisager au niveau clinique, biologique et radiologique.4
Cliniquement, il faut évoquer une HPT1 devant des douleurs diffuses, des lithiases rénales, une ostéoporose, des fractures répétées, des troubles cognitifs, parfois psychiatriques, ou des troubles de la conscience.Néanmoins, le diagnostic différentiel de l’HPT1 est surtout biologique, en particulier dans les formes atypiques, qui doivent être dissociées :
- des hypercalcémies avec hypocalciurie ou PTH non accrue ;
- des situations de normocalcémie avec PTH élevée, hypophosphatémie ou hypercalciurie (tableaux 2 et 3).
Conclure à un diagnostic différentiel nécessite une analyse préalable des facteurs susceptibles de perturber les paramètres phosphocalciques : carence en vitamine D, insuffisance rénale, malabsorption, insuffisance d’apports calciques et causes iatrogènes.
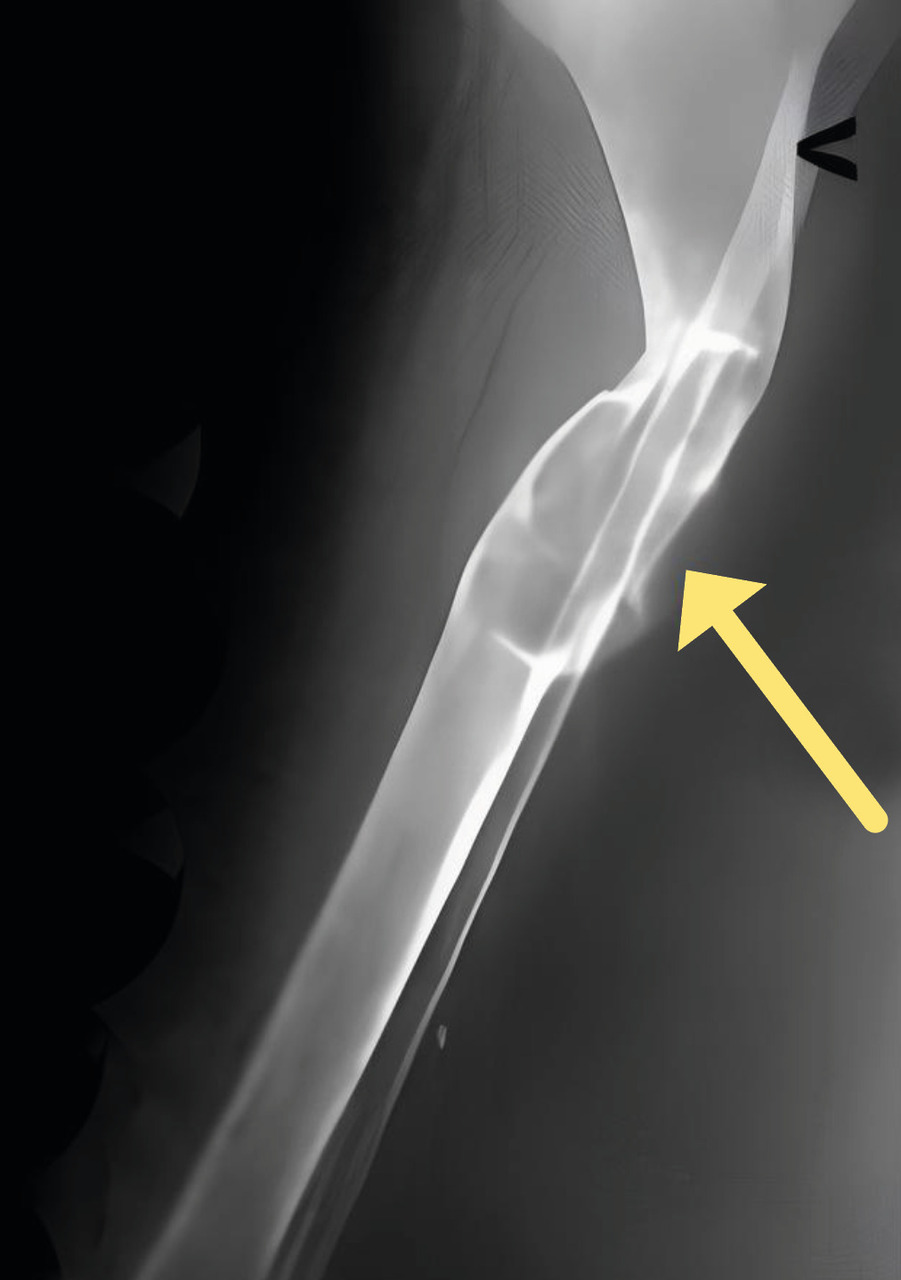
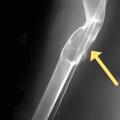
Sur le plan radiologique, une tumeur brune (fig. 2), devenue rare, doit être différenciée principalement d’une métastase osseuse, d’un chondrosarcome ou d’une tumeur à cellules géantes.
Formes sporadiques et génétiques
Une fois le diagnostic biologique affirmé, une forme génétique (environ 10 % des HPT1) doit être évoquée : soit syndromique (néoplasies endocriniennes multiples [NEM] de type 1, 2A et 4 ; syndrome HPT1 -tumeur mandibulaire), soit familiale isolée.
La reconnaissance de ces formes génétiques est importante afin de garantir une prise en charge adaptée au gène et au type de variant impliqué (exploration chirurgicale d’emblée bilatérale si décision opératoire) avec présence d’un variant génétique. Cependant, la recherche d’une cause génétique n’est pas justifiée chez l’ensemble des patients avec HPT1 ;5 elle l’est en cas de forme familiale d’HPT1 (au moins deux apparentés au 1er ou 2e degré), de survenue avant l’âge de 50 ans, ou après 50 ans mais avec une forme récidivante ou multiglandulaire, ou suspecte de malignité.
À ce jour, il est recommandé d’explorer en première intention le panel de gènes suivant : MEN1, CDKN1B, CDC73, CASR, GNA11, AP2S1 et GCM2. En pratique, les situations les plus fréquentes sont les NEM de type 1 associant HPT1 multiglandulaire survenant chez un sujet jeune, tumeur neuro-endocrine du pancréas et adénome hypophysaire.
La néoplasie endocrinienne multiple de type 2 associant phéochromocytome, cancer médullaire de la thyroïde et HPT1 est, à l’inverse, rarement révélée par l’HPT1, mais sa suspicion nécessite un dosage préopératoire de la calcitonine et des catécholamines.
Bilan d’imagerie de l’HPT1
L’HPT1 est la conséquence d’une hypersécrétion de PTH par un adénome parathyroïdien unique dans plus de 80 % des cas. Une atteinte multiglandulaire, associant dans des proportions variables adénome et/ou hyperplasie, est également possible, quoique moins fréquente.
Lorsque le diagnostic d’HPT1 est certain et qu’un geste chirurgical est envisagé, l’imagerie est utile pour localiser la ou les glande(s) hyperfonctionnelle(s).
La stratégie d’exploration repose en première intention sur l’association de deux examens :
- une échographie parathyroïdienne ;
- un examen de médecine nucléaire, pouvant être une scintigraphie parathyroïdienne, de préférence en double isotope 123I/99mTc-Sestamibi, avec acquisitions planaires et tomoscintigraphiques, ou une TEP-TDM à la 18F-choline.7 En cas de résultats négatifs, il est conseillé de réaliser une TEP 18F-choline si le premier examen était une scintigraphie et de bien explorer le médiastin, parfois siège d’adénome parathyroïdien ectopique.
Dans les situations difficiles, un scanner parathyroïdien en 4D ou une IRM parathyroïdienne, une cytoponction avec dosage de PTH dans le liquide de rinçage sont possibles après discussion pluridisciplinaire en centre expert, d’autant que le scanner parathyroïdien est assez irradiant et ne doit pas être réitéré indûment.
Prise en charge de l’HPT1 : approche par organes cibles
Dans le contexte d’une HPT1, l’eucalcémie est souvent un critère intermédiaire et ne doit pas être utilisée comme un objectif thérapeutique en soi.8,9 En effet, la diminution de la calcémie témoigne de l’efficacité mécanistique du traitement mais ne mesure pas le bénéfice potentiel apporté au patient. Ainsi, chez le sujet âgé ayant une hypercalcémie modérée, il n’est pas toujours évident que le traitement apporte aussi un bénéfice en matière de symptômes, de qualité de vie, de troubles cognitifs, de complications (cardiovasculaires ou autres), voire de risque de décès. Cela justifie, dans la plupart des recommandations récentes, l’utilisation de l’approche par « organe cible », en distinguant les patients avec ou sans atteinte potentiellement attribuable à l’HPT1 au moment du diagnostic.
Les patients hypercalcémiques doivent bien s’hydrater (2 à 3 litres d’eau par jour, surtout en période caniculaire) et il faut veiller à ce que l’apport calcique soit préservé afin d’éviter une ascension de la PTH par carence calcique qui aggraverait l’ostéoporose et l’hypercalcémie. Toute hypercalcémie sévère nécessite une prise en charge spécifique détaillée ci-après, en particulier avant une prise en charge chirurgicale.
Préparation à l’intervention
En dehors du bilan étiologique, notamment génétique si nécessaire, le traitement d’une hypercalcémie sévère évoluant dans le cadre d’une HPT1 a deux objectifs :
- prise en charge d’une hypercalcémie sévère et/ou symptomatique ;
- prévention d’une hypocalcémie postopératoire.
L’hypercalcémie sévère, définie par une calcémie supérieure ou égale à 3,5 mmol/L, nécessite une admission dans un service d’hospitalisation conventionnelle ou en soins critiques, selon son retentissement et les comorbidités.10 L’admission en soins critiques s’appuie sur l’existence d’une ou de plusieurs manifestations cliniques (troubles de la vigilance, déshydratation avec insuffisance rénale aiguë, pancréatite aiguë sévère), de signes électrocardiographiques menaçants, d’une ou de plusieurs comorbidités importantes, notamment cardiovasculaires.
Une réhydratation par voie orale et/ou une expansion volémique par voie intraveineuse, adaptée à la fonction cardiaque et rénale, constituent la base du traitement.
Les bisphosphonates (zolédronate ou pamidronate), administrés en intraveineux, sont également recommandés en première intention pour obtenir une baisse de la calcémie suffisamment longue pour organiser la chirurgie. Toute injection de bisphosphonate doit être précédée d’un bilan étiologique minimal de l’hypercalcémie comportant un dosage sanguin de PTH, de phosphates, de 25 -hydroxyvitamine D et une calciurie et créatininurie au moins sur un échantillon, car les bisphosphonates modifient durablement ces paramètres. Puisque les bisphosphonates agissent après un délai de vingt-quatre à trente-six heures, la calcitonine peut être initialement associée en raison de son action rapide, en quelques heures.
La prescription de cinacalcet peut être discutée, mais il n’y a pas de consensus dans la littérature, le risque étant de retarder indûment la chirurgie chez un patient qui a déjà eu une hypercalcémie sévère.
Le dénosumab est recommandé en deuxième intention, si les bisphosphonates ne peuvent pas être utilisés, notamment du fait d’une altération de la fonction rénale.
L’épuration extrarénale est réservée aux patients ayant un risque vital identifié, surtout si une expansion volémique n’est pas envisageable en raison d’une insuffisance cardiaque ou rénale.
La correction de la carence en vitamine D est recommandée avant la chirurgie parathyroïdienne dès lors que la calcémie est inférieure à 3,5 mmol/L pour prévenir ou atténuer l’hypocalcémie postopératoire sévère par transfert massif de calcium dans l’os.
Chirurgie
L’approche chirurgicale sélective (c’est-à-dire ciblée sur la lésion) est recommandée pour les patients avec une seule glande hyperfonctionnelle à l’imagerie (morphologique et fonctionnelle).
L’utilisation des dosages peropératoires de la PTH peut réduire les taux d’échec.
Si une maladie multiglandulaire est découverte, si aucun adénome parathyroïdien n’est visualisé, ou si la concentration peropératoire de PTH ne diminue pas de manière appropriée après l’excision de l’adénome suspecté, le chirurgien doit envisager une conversion pour réaliser une exploration bilatérale.
L’exploration cervicale bilatérale des quatre glandes est recommandée d’emblée en cas de suspicion de maladie multiglandulaire, d’imagerie préopératoire non contributive, dans les formes héréditaires d’HPT1, dans les formes induites par le lithium, ou lorsque la décroissance peropératoire de la concentration de la PTH n’est pas satisfaisante.
L’autofluorescence et le neuromonitorage peropératoire du nerf laryngé inférieur peuvent aider à éviter une complication récurrentielle. L’autre risque de cette chirurgie est la survenue d’un hématome cervical postopératoire, dont la prise en charge immédiate est essentielle.
Les indications opératoires figurent dans le tableau 4.
Traitements médicamenteux
Les traitements médicamenteux de l’HPT1 relèvent de plusieurs classes thérapeutiques : calcimimétiques (cinacalcet), bisphosphonates (alendronate, risédronate, ibandronate, zolédronate) – qui protègent surtout de l’ostéoporose –, anticorps monoclonal inhibiteur des ostéoclastes (dénosumab), œstrogènes, calcium et vitamine D.
Un traitement pharmacologique peut être proposé dans les situations suivantes :
- indication opératoire mais refus de la chirurgie par le patient ;
- chirurgie non indiquée, après évaluation multidisciplinaire de la balance bénéfices/risques ;
- maladie persistante après une exploration chirurgicale par une équipe entraînée, au cas par cas et après discussion multidisciplinaire.
- Chez ces patients, quatre modalités thérapeutiques sont recommandées :
- un apport en calcium (800 mg/j pour les femmes de moins de 50 ans et les hommes de moins de 70 ans ; 1 000 mg/j pour les femmes de plus de 50 ans et les hommes de plus de 70 ans) ;
- un objectif de concentration plasmatique en vitamine D supérieur à 30 ng/mL ;
- un traitement par cinacalcet si la calcémie est supérieure de 0,25 mmol/L par rapport à la limite haute de la norme (faible niveau de preuve) ;
- un traitement par bisphosphonates s’il existe une ostéoporose densitométrique.
Aucun traitement médicamenteux n’a montré de réelle efficacité sur la diminution des lithiases rénales. Des conseils diététiques de prévention de la lithogenèse, identiques à ceux destinés aux patients sans HPT1, sont donc primordiaux. Les diurétiques thiazidiques permettant de diminuer la calciurie pourraient être proposés dans cette indication, mais du fait de leur effet hypercalcémiant, aucune étude n’a été réalisée à ce jour dans l’HPT1. La supplémentation prudente et raisonnable en vitamine D n’a pas d’effet sur les niveaux de calciurie mais peut protéger l’os.
Le traitement hormonal substitutif œstrogénique permet la réduction du turnover osseux, avec amélioration de la densitométrie osseuse et réduction de la calciurie chez les femmes ménopausées avec HPT1. Il n’y a cependant pas de données spécifiques sur la réduction du risque de fractures osseuses dans l’HPT1.
Destruction locale
Le traitement par destruction locale (thermoablation) doit être réalisé dans un service spécialisé, après validation en réunion de concertation pluridisciplinaire. Les éléments de la discussion concernent l’accessibilité d’une cible échographique, l’expertise de l’opérateur, l’acceptation d’un risque récurrentiel et de la nécessité d’un suivi biologique et échographique.
À l’heure actuelle, la destruction locale ne semble recommandée qu’en cas d’échec, d’intolérance ou de refus du traitement médical ou chirurgical.
Pronostic et modalités de surveillance
Un dosage de la PTH effectué dans les vingt-quatre heures postopératoires supérieur à 10 pg/mL permet d’identifier les patients qui ne développeront pas d’hypoparathyroïdie permanente.
Si l’HPT1 est traitée chirurgicalement, une surveillance étroite de la calcémie est nécessaire en postopératoire pour détecter la survenue d’une hypocalcémie.11
L’hypocalcémie peut être provoquée par une hypoparathyroïdie (notamment en cas de chirurgie multiglandulaire ou de reprise chirurgicale) ou par le syndrome de l’os avide, complication rare se manifestant par l’installation d’une hypocalcémie et d’une hypophosphatémie sévère (tableau 5). Cette dernière doit être suspectée en cas de lésions osseuses importantes ou de carence préopératoire sévère en vitamine D.
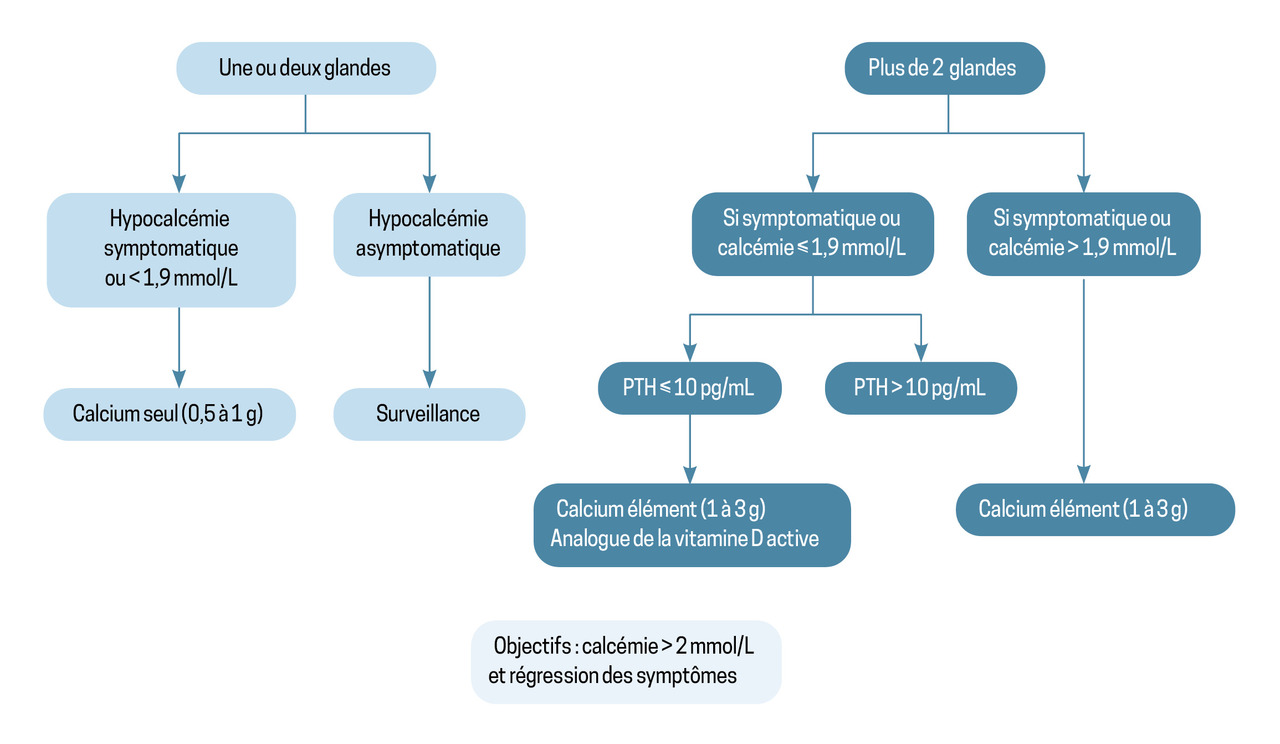
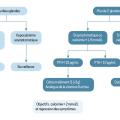
Le traitement de l’hypocalcémie postopératoire dépend de la gravité, des symptômes et de l’intervention chirurgicale réalisée (figure 3). Si ce traitement est insuffisant, l’intérêt d’une supplémentation par vitamine D active (calcitriol) est évalué et le suivi est assuré en collaboration avec un endocrinologue (calcémie, calciurie et éventuellement phosphatémie et PTH).
Sous traitement médical de l’hypoparathyroïdie, la calcémie doit être surveillée au moins tous les trois mois la première année, puis au moins deux fois par an.
La nouvelle classification des tumeurs parathyroïdiennes de l’Organisation mondiale de la santé datant de 2022 (5e édition) souligne les caractéristiques histologiques potentiellement indicatrices d’anomalies génétiques sous-jacentes en raison de leur implication dans la prise en charge des patients.
Le carcinome parathyroïdien est d’une extrême rareté (moins de 1 % des HPT1). Pour cette raison, sa prise en charge doit être effectuée en centre expert.
Une HPT1 persistante est définie par la persistance ou la récidive d’une hypercalcémie dans les six mois qui suivent une chirurgie parathyroïdienne. Une HPT1 récidivante est définie par la réapparition d’un tableau biologique d’HPT1 plus de six mois après une parathyroïdectomie initialement curative. Ces situations nécessitent une prise en charge dans un centre expert.
Depuis la publication des premières recommandations portées par la Société française d’endocrinologie en 2006, aucun document de référence n’avait fait l’objet d’un travail consensuel sur les HPT1. Une synthèse de ces évolutions paraissait utile, et la conduite à tenir devant une suspicion d’HPT1 est résumée dans la fig. 4.
Cas particuliers
Chez l’enfant
L’HPT1 est rare et d’origine génétique dans la moitié des cas chez l'enfant. Les manifestations cliniques sont très variables (souvent absentes chez le nouveau-né, plutôt sévères et symptomatiques chez l’enfant et l’adolescent). L’exploration morphologique et le traitement suivent les mêmes recommandations que celles de l’adulte. La chirurgie doit être réalisée par un chirurgien référent.6
Chez la femme enceinte
L’HPT1 est rarement diagnostiquée au cours de la grossesse car les modifications de l’homéostasie phosphocalcique liées à la grossesse peuvent en masquer la symptomatologie (vomissements). La morbidité materno-fœtale associée à l’HPT1 pendant la grossesse, historiquement considérée comme importante, est en réalité rare et dépend de la calcémie maternelle. Le traitement (conservateur ou chirurgical) doit être adapté au terme de la grossesse, à la sévérité des symptômes et aux risques materno-fœtaux.6
Chez le sujet âgé
L’HPT1 est fréquente chez le sujet âgé. Bien que la démarche diagnostique soit comparable à celle des patients plus jeunes, la forte prévalence des fractures ostéoporotiques et de leurs conséquences en matière de morbi-mortalité chez les sujets âgés doit conduire à privilégier la prise en charge chirurgicale en cas d’ostéoporose. Les symptômes neuropsychologiques et l’atteinte cardiovasculaire de l’HPT1 ne justifient pas de recommander la parathyroïdectomie. Le recours à la chirurgie reste très insuffisant, en comparaison à la population plus jeune, alors qu’elle est le traitement de première intention, et que sa sécurité et son efficacité sont largement démontrées. L’indication opératoire peut être guidée par les critères de fragilité de Fried (au moins 3 critères parmi les suivants : perte de poids involontaire ≥ 5 % en un an, fatigue subjective, diminution de l’activité physique, vitesse de marche lente, diminution de la force de préhension) mais aussi par l’autonomie et les comorbidités. Si l’indication opératoire n’est pas retenue, un apport vitaminocalcique régulier, un traitement antiostéoporotique si besoin et du cinacalcet (si la calcémie est supérieure à 2,75 mmol/L) sont indiqués.6
Que dire à vos patients ?
L’hyperparathyroïdie primaire est une pathologie assez fréquente, surtout chez la femme ménopausée.
Dans de très rares cas, elle peut survenir avant l’âge de 40 ans ; il faut alors rechercher des formes familiales par un test génétique.
L’hyperparathyroïdie est presque toujours liée à un adénome parathyroïdien, petite tumeur bénigne touchant l’une des quatre glandes parathyroïdes. Elle peut entraîner une fragilité osseuse, des fractures, des calculs rénaux, une insuffisance rénale, parmi les conséquences les plus fréquentes.
La prise en charge chirurgicale est préférable s’il existe un retentissement, chez les personnes jeunes ou si la calcémie est supérieure à 2,75 mmol/L, après un bilan de localisation soigneux et par un chirurgien expert compte tenu du risque d’atteinte récurrentielle et d’hypoparathyroïdie dans les explorations bilatérales du cou.
2. Bouillet B, Bertocchio JP, Nomine-Criqui C, et al. Chapter 2: Primary hyperparathyroidism: Diagnosis. Ann Endocrinol (Paris) 2025;86(1):101691.
3. Scheyer N, Frey S, Koumakis E, et al. Chapter 3: Impact of primary hyperparathyroidism. Ann Endocrinol (Paris) 2025;86(1):101692.
4. Kamenický P, Houillier P, Vantyghem MC. Chapter 4: Differential diagnosis of primary hyperparathyroidism. Ann Endocrinol (Paris) 2025;86(1):101693.
5. Al-Salameh A, Haissaguerre M, Tresallet C, et al. Chapter 6: Syndromic primary hyperparathyroidism. Ann Endocrinol (Paris) 2025;86(1):101695.
6. Lemaitre M, Picart C, Gueorguieva I, et al. Chapter 7: The different forms of primary hyperparathyroidism at different ages of life: Childhood, pregnancy, lactation, old age. Ann Endocrinol (Paris) 2025;86(1):101696.
7. Chevalier B, Ghander C, Ladsous M, et al. Chapter 10: What parathyroid imaging is required for hyperparathyroidism? Ann Endocrinol (Paris) 2025;86(1):101699.
8. Frey S, Mosbah H, Donatini G, et al. Chapter 9: Indications for the treatment of primary hyperparathyroidism. Ann Endocrinol (Paris) 2025;86(1):101698.
9. Baud G, Espiard S, Buffet C, et al. Chapter 11: Treatment modalities. Ann Endocrinol (Paris) 2025;86(1):101700.
10. Lecoq AL, Jannin A, Cirenei C, et al. Chapter 12: Preparation for parathyroid surgery. Ann Endocrinol (Paris) 2025;86(1):101701.
11. Barraud S, Lopez AG, Sokol E, et al. Chapter 14: Post surgical follow-up of primary hyperparathyroidism. Ann Endocrinol (Paris) 2025;86w (1):101703.
Dans cet article
 Encadrés
Encadrés



L'hyperparathyroïdie est souvent découverte sur une hypercalcémie lors d’un bilan de routine. Elle nécessite une confirmation diagnostique démontrant l’hypophosphatémie, l’hypercalciurie, la normalité de la créatininémie et l’augmentation de la PTH.
Pour que le bilan biologique de confirmation (et notamment le dosage de la PTH) soit interprétable, il faut idéalement arrêter les diurétiques et avoir un taux de vitamine D normal, vérifier l’absence de traitement par bisphosphonates (même annuels) ou par lithium.
Une hyperparathyroïdie doit être recherchée devant des fractures, des tassements vertébraux, une ostéoporose, des coliques néphrétiques.
Même si l’hypercalcémie est modérée, un bilan du retentissement est à effectuer par un endocrinologue afin de poser ou non l’indication opératoire.
En cas d’indication opératoire, le bilan de localisation est réalisé, en commençant par une échographie parathyroïdienne, les autres examens étant demandés par l’endocrinologue.