L’hypogonadisme masculin correspond à une diminution de la production d’hormones sexuelles mâles, essentiellement la testostérone. La production de testostérone est sous la dépendance de la pulsatilité de la gonadolibérine (GnRH) hypothalamique, puis de l’hormone lutéinisante (LH) hypophysaire (fig. 1). La carence en testostérone peut se manifester par une variété de symptômes, tels qu’un micropénis détecté à la naissance ou un impubérisme, mais surtout par des signes souvent peu spécifiques, comme une fatigue, des bouffées de chaleur, des sueurs, une diminution de la masse musculaire, une augmentation de la masse grasse, une baisse de la libido, des troubles érectiles, une infertilité ou encore une ostéopénie, voire une ostéoporose.1 Sur le plan biologique, l’hypogonadisme se définit par des signes cliniques associés à un taux de testostérone totale le matin inférieur à 3 ng/L (10 nmol/L) chez l’adulte, soit deux écarts types en dessous de la moyenne d’une population de même âge.2
La démarche devant un hypogonadisme est initialement de trouver son origine, qui peut être centrale (hypothalamique et/ou hypophysaire) ou périphérique (testiculaire). Le traitement n’est à envisager qu’après avoir identifié la cause de l’hypogonadisme.
La prévalence exacte de l’hypogonadisme masculin est mal connue et probablement sous-estimée. Un hypogonadisme est présent chez environ 1 homme adulte sur 200, et ce chiffre augmente après l’âge de 60 ans. Selon une grande étude européenne (European male ageing study), la prévalence atteindrait même 2 à 3 % de la population masculine.3
Confirmation diagnostique et orientation étiologique
Le dosage initial recommandé est celui de la testostérone totale – idéalement le matin à jeun, en raison de la cyclicité de la testostérone.
Un taux de testostérone totale inférieur à 1 ng/mL (3,56 nmol/L) affirme le diagnostic d’hypogonadisme.
Si le taux se situe entre 1 et 3 ng/mL, un deuxième prélèvement est souhaitable.
Le dosage de la testostérone libre est déconseillé en raison de son manque de fiabilité analytique.
Dans certains cas, comme dans l’obésité sévère, le dosage de la globuline liant les hormones sexuelles (SHBG) permet de calculer la testostérone biodisponible : cette dernière correspond à la fraction non liée à la SHBG et comprend la fraction libre et liée à l’albumine.
La mesure concomitante des taux de LH et de l’hormone de stimulation folliculaire (FSH) permet de distinguer les formes centrales (hypogonadotropes) des formes périphériques (hypergonadotropes).
L’examen clinique comprend l’évaluation de la fréquence du rasage, de la masse musculaire et le calcul de l’indice de masse corporelle.
La palpation testiculaire se fait à l’aide d’un orchidomètre. La taille testiculaire normale est d’environ 15 à 20 mL. Une échographie testiculaire peut être préconisée pour obtenir une mesure plus précise.
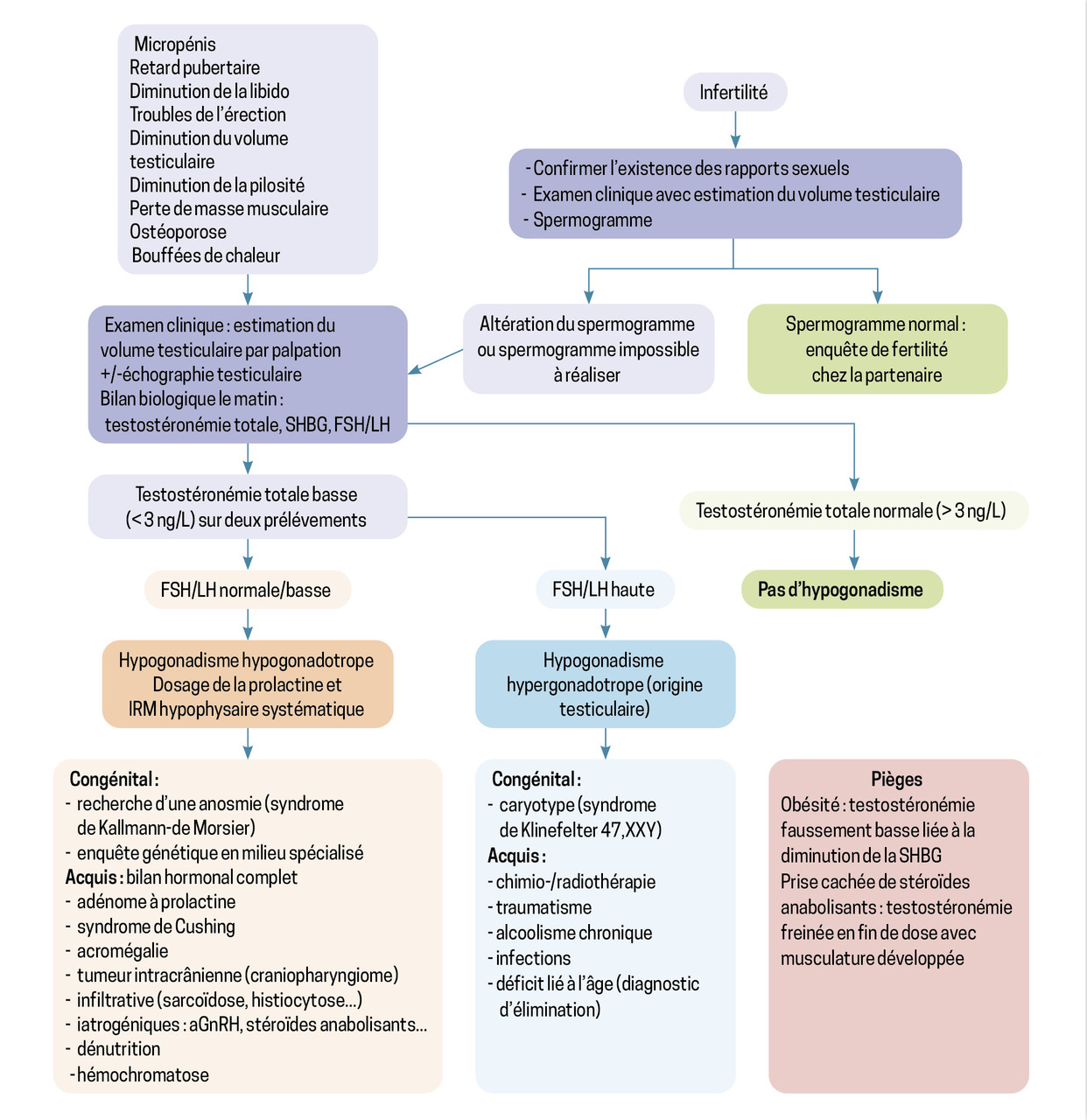
Un algorithme diagnostique devant des signes cliniques d’hypogonadisme ou d’infertilité est proposé en figure 2.
Hypogonadismes hypogonadotropes (origine centrale)
En cas d’hypogonadisme hypogonadotrope (HH), la LH et la FSH sont basses ou anormalement normales malgré un taux de testostérone bas. Dans ces situations, une IRM hypothalamo-hypophysaire est nécessaire.
Forme congénitale
L’HH congénital (HHC) est rare. Il peut être diagnostiqué à la naissance devant un micropénis et/ou une cryptorchidie. Mais, le plus souvent, il est diagnostiqué à l’adolescence, devant un retard pubertaire (défini par l’absence d’augmentation du volume testiculaire après l’âge de 14 ans et un défaut de croissance), un micro-pénis (défini par une taille en érection inférieure à 7 cm), une cryptorchidie et/ou une faible pilosité. Il est important de noter que la pilosité pubienne n’est pas un signe de puberté mais le reflet d’une sécrétion d’androgènes surrénaliens.4
Le déficit en androgènes est généralement plus sévère dans les formes congénitales que dans les formes acquises, avec une testostéronémie très basse et un volume testiculaire plus faible.
L’interrogatoire s’attache à rechercher d’autres cas d’hypogonadisme dans la famille. La présence d’un trouble de l’olfaction oriente vers un syndrome de Kallmann-de Morsier.4
Une analyse génétique est recommandée en cas d’HHC. Elle doit être réalisée en milieu spécialisé. Plus d’une cinquantaine de gènes ont été identifiés à ce jour comme étant impliqués dans des HHC. L’analyse par séquençage à haut débit permet actuellement de trouver un variant génétique pathogène dans environ 50 % des cas. Un conseil familial est nécessaire en cas de découverte d’un tel variant.
Forme acquise
L’hypogonadisme hypogonadotrophique peut survenir à tout âge dans sa forme acquise. Il s’agit de tableaux généralement moins sévères que dans les formes congénitales et dont le phénotype est très variable.
Un bilan hormonal hypophysaire complet est indispensable pour exclure une cause endocrinienne (hyperprolactinémie, hypercortisolisme, acromégalie...). Une hyperprolactinémie peut être secondaire à une prise médicamenteuse, à une insuffisance rénale sévère ou à un adénome hypophysaire.
En cas d’HH acquis, l’IRM hypophysaire est indispensable, pour ne pas méconnaître une atteinte tumorale (adénome, craniopharyngiome…) ou infiltrative (sarcoïdose, histiocytose). Il est aussi nécessaire d’évoquer une hémochromatose. En effet, cette pathologie génétique peut se manifester par un hypogonadisme acquis par accumulation de fer dans les cellules gonadotropes hypophysaires.
Il convient également de penser aux causes iatrogéniques. L’utilisation d’agonistes de la GnRH dans les cancers de la prostate est ainsi une cause classique d’hypogonadisme acquis. Par ailleurs, un usage détourné de stéroïdes anabolisants – autres que la testostérone – dans un contexte sportif est possiblement en cause ; une testostéronémie basse, avec un taux de gonadotrophines bas, associée à un phénotype de musculature bien développée doit faire envisager ce mésusage.
Enfin, le diagnostic d’hypogonadisme hypothalamique d’origine fonctionnelle (dénutrition ou sélection alimentaire avec peu de lipides) est possible chez l’homme. Il survient lorsque la balance énergétique est négative, parfois dans un contexte d’activité sportive intense. Le diagnostic ne peut être établi qu’après avoir éliminé les causes organiques.
Hypogonadismes hypergonadotropes ou insuffisance testiculaire
Des taux élevés de FSH et de LH, supérieurs à la normale, reflètent une atteinte testiculaire. Les taux de testostéronémie sont généralement moins diminués que dans l’hypogonadisme hypogonadotrope.
Forme congénitale : surtout le syndrome de Klinefelter
Le diagnostic le plus fréquent est le syndrome de Klinefelter, qui concerne 1 garçon sur 600. Il est parfois diagnostiqué en pédiatrie. Des études épidémiologiques, notamment au Danemark, ont mis en évidence un sous-diagnostic important, avec moins de 30 % des cas identifiés au cours de la vie.5 Auparavant, de nombreux diagnostics étaient réalisés lors de l’examen clinique du service militaire. À l’heure actuelle, beaucoup sont révélés dans le cadre d’une infertilité avec azoospermie.Le syndrome de Klinefelter est diagnostiqué lorsqu’il existe un chromosome X surnuméraire. Le caryotype – obtenu après un consentement éclairé – revient 47,XXY ou sous une forme mosaïque 46,XY/47,XXY. Le phénotype correspond classiquement à un garçon de grande taille, avec de longs membres.
Ce syndrome expose à un surrisque de diabète de type 2, d’hypothyroïdie et de cancer du sein. La puberté survient le plus souvent à un âge normal, mais les testicules restent de petite taille, la pilosité est faible et s’accompagne fréquemment d’une gynécomastie. Cette pathologie est aussi à l’origine d’apnées du sommeil et d’arthrose. Il peut exister une baisse des acquisitions, avec en moyenne 10 à 15 points de QI de moins que les autres membres de la famille. L’existence de troubles du comportement dans l’enfance ou à l’adolescence à type d’agitation est un mode fréquent de découverte et doit mener à un accompagnement du patient et de sa famille afin de favoriser la scolarité et la future insertion professionnelle. Toutefois, le diagnostic peut passer inaperçu.
Il existe d’autres causes chromosomiques ou génétiques beaucoup plus rares : hommes XX, maladie de Steinert, syndrome de Noonan, etc.
Formes acquises
Les causes acquises sont à rechercher à l’interrogatoire et sont principalement iatrogéniques par chimiothérapie (notamment les agents alkylants) ou radiothérapie, liées à un traumatisme testiculaire, ou à une castration ou une torsion testiculaire prise en charge tardivement.
L’alcoolisme chronique est probablement l’une des causes d’hypogonadisme les plus fréquentes, mais le diagnostic est souvent difficile en raison de l’augmentation de la SHBG secondaire à l’hépatopathie alcoolique, responsable de taux de testostérone faussement normaux.
Les causes infectieuses, comme l’orchite secondaire au virus des oreillons, sont devenues très rares depuis la généralisation de la vaccination. Une atteinte bilatérale est alors observée. Une forme secondaire à une infection par le gonocoque est aussi possible.
Le diagnostic de déficit androgénique lié à l’âge est un diagnostic d’élimination. Il ne doit être considéré que chez des patients de plus de 55 à 60 ans ayant un taux de testostérone inférieur à 3 ng/L (10 nmol/L), avec un retentissement clinique significatif. Certains signes sont non spécifiques et peuvent être confondus avec ceux du vieillissement normal. Le diagnostic est porté sur le taux de testostérone totale. En cas d’hypogonadisme franc, d’autant plus que les taux de gonadotrophines sont bas, il convient de ne pas méconnaître un autre diagnostic, notamment une pathologie hypophysaire.
Modalités de la substitution hormonale
Le traitement substitutif par testostérone peut être initié en médecine générale ou en collaboration avec un endocrinologue ou un urologue. En France, la substitution hormonale est disponible sous forme de gels transdermiques et par injection intramusculaire (tableau). Les comprimés et patchs ne sont plus disponibles depuis 2025.
L’adaptation du traitement dépend de la clinique et du contrôle de la testostéronémie totale.
Contre-indications
Les contre-indications absolues au traitement substitutif par testostérone sont les suivantes :
-
polyglobulie avec un taux d’hématocrite supérieur à 55 % du fait du risque cardiovasculaire ;
-
cancer androgénodépendant (de la prostate ou du sein) ; à noter que le traitement par testostérone ne favorise pas la survenue d’un cancer de la prostate, mais ce cancer étant hormonodépendant, le traitement ne doit pas être prescrit si celui-ci préexiste ;6 toutefois, un traitement peut être envisagé en cas de cancer de la prostate opéré, à faible risque et en rémission, après un avis spécialisé ;7
-
hypercalcémie associée à une tumeur maligne ;
-
tumeur hépatique ou antécédent de tumeur hépatique ;
-
insuffisance cardiaque, hépatique ou rénale sévère.
Précautions d’emploi
La substitution androgénique doit être utilisée avec prudence chez les patients ayant une pathologie cardiaque, hépatique ou rénale.
Les données sont controversées concernant les effets des androgènes chez l’homme de plus de 60 ans ayant une pathologie cardiovasculaire, même si les dernières études sont plutôt rassurantes.
Des précautions sont également nécessaires en cas de thrombophilie ou de facteurs de risque de maladie thrombo-embolique veineuse : dans ces situations, un avis spécialisé est préconisé.
Quelle surveillance ?
La surveillance nécessite une évaluation clinique et biologique tous les trois mois la première année, puis annuelle. L’interrogatoire porte sur la libido, les érections, la fonction sexuelle, les bouffées de chaleur. La surveillance clinique inclut un contrôle du poids, de l’indice de masse corporelle, de la pression artérielle et de la masse musculaire.
Le bilan biologique comprend :
-
un hémogramme, avec surveillance de l’hématocrite (arrêt temporaire du traitement si son taux dépasse 55 %) ;
-
un dosage sanguin du PSA (voire un toucher rectal selon l’âge du patient) ;
-
un bilan lipidique complet avec cholestérol total, HDL, LDLc, triglycérides ;
-
la glycémie à jeun ainsi que le taux d’hémoglobine glyquée si le patient est diabétique.
En outre, une ostéodensitométrie – à réaliser environ tous les cinq ans – permet de surveiller la masse osseuse.
Enfin, une évaluation du retentissement psychologique de la maladie et de la prise en charge est nécessaire ; un soutien est à proposer si besoin.
Pronostic, évolution et fertilité
Le pronostic en matière de fertilité est étroitement lié à la cause de l’hypogonadisme. Il est important de rassurer le patient : l’hypogonadisme n’est pas obligatoirement synonyme d’infertilité.
La substitution en testostérone, en raison d’un potentiel surdosage, peut inhiber la spermatogenèse par freination potentielle de l’axe hypothalamo-hypophysaire ; elle est suspendue en cas de projet de paternité.
Hypogonadisme central
Le pronostic de fertilité dépend essentiellement de l’âge au diagnostic. Il est plus sévère en cas d’hypogonadisme congénital qu’en cas d’hypogonadisme acquis (ou secondaire) où une puberté classique a permis un développement testiculaire satisfaisant.
En cas d’hypogonadisme central acquis, le traitement de la cause peut permettre une restauration de l’axe hypothalamo-hypophysaire, une normalisation des taux hormonaux et une récupération de la spermatogenèse spontanée.8 Par exemple, un traitement par agonistes dopaminergiques dans le cadre d’un adénome à prolactine peut permettre de rétablir la spermatogenèse.
Dans les autres cas, l’endocrinologue, l’urologue ou un médecin de centre d’assistance médicale à la procréation (AMP) peut induire la spermatogenèse à l’aidede traitements comportant deux gonadotrophines pour remplacer la FSH et la LH. Ce traitement est long (environ 12 à 24 mois), à raison de trois injections par semaine. La surveillance se fait par des dosages réguliers de testostérone et d’inhibine B. Généralement, le spermogramme est demandé après quatre à cinq mois de traitement au minimum.
Hypogonadisme périphérique
Le pronostic de fertilité est évalué sur le spermogramme. Si ce dernier est altéré, une prise en charge spécialisée en AMP doit être proposée, avec éventuellement une injection cytoplasmique de spermatozoïdes (ICSI) directement dans l’ovocyte. Si le spermogramme est très altéré ou en cas d’azoospermie, un don de gamètes doit être proposé. Pour certains patients avec hypogonadisme d’origine testiculaire, une biopsie testiculaire peut permettre l’obtention de gamètes viables malgré une azoospermie. Par exemple, pour les patients atteints du syndrome de Klinefelter, l’obtention de spermatozoïdes est possible dans 40 à 50 % des cas.9 Il s’agit de la technique appelée TESE (Testicular Sperm Extraction).
Cas particulier de la prise en charge en pédiatrie
La prise en charge de l’hypogonadisme masculin pédiatrique doit se faire en milieu spécialisé. Une induction pubertaire est initiée par les endocrinologues pédiatres. La substitution par testostérone chez l’enfant se fait à dose croissante. Ce traitement induisant une maturation du cartilage de conjugaison, le pronostic de croissance de l’enfant est engagé et une surveillance spécialisée est requise.
Que dire à vos patients ?
L’hypogonadisme masculin correspond à la production insuffisante de testostérone par les testicules.
Le traitement hormonal substitutif permet une amélioration des signes cliniques (baisse de la libido, masse musculaire, fatigue, etc.) dans l’immense majorité des cas.
Hypogonadisme ne rime pas avec infertilité. Le spermogramme est l’examen de première intention pour évaluer les possibilités de fertilité.
2. Bhasin S, Cunningham GR, Hayes FJ, et al. Testosterone therapy in men with androgen deficiency syndromes: An Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2010;95(6):2536-59.
3. Tajar A, Huhtaniemi IT, O’Neill TW, et al. Characteristics of androgen deficiency in late-onset hypogonadism: Results from the European male aging study (EMAS). J Clin Endocrinol Metab 2012;97(5):1508-16.
4. Young J. Approach to the male patient with congenital hypogonadotropic hypogonadism. J Clin Endocrinol Metab 2012;97(3):707-18.
5. Bojesen A, Juul S, Gravholt CH. Prenatal and postnatal prevalence of Klinefelter syndrome: A national registry study. J Clin Endocrinol Metab 2003;88(2):622-6.
6. Baik SH, Baye F, Fung KW, et al. Association between testosterone replacement therapy and prostatic disorders in elderly hypogonadal men. J Clin Endocrinol Metab 2026;111:794-810.
7. Santucci J, Stapleton P, Perera M, et al. Oncological safety of testosterone replacement therapy in men with localised prostate cancer: A systematic review of observational studies. BJU Int 2025;136(5):788-99.
8. Papadakis GE, de Kalbermatten B, Dormoy A, et al. Impact of Cushing’s syndrome on the gonadotrope axis and testicular functions in men. Hum Reprod 2023;38(12):2350-61.
9. Corona G, Pizzocaro A, Lanfranco F, Garolla A, et al. Sperm recovery and ICSI outcomes in Klinefelter syndrome: A systematic review and meta-analysis. Hum Reprod Update 2017;23:265-75.
Dans cet article
 Encadrés
Encadrés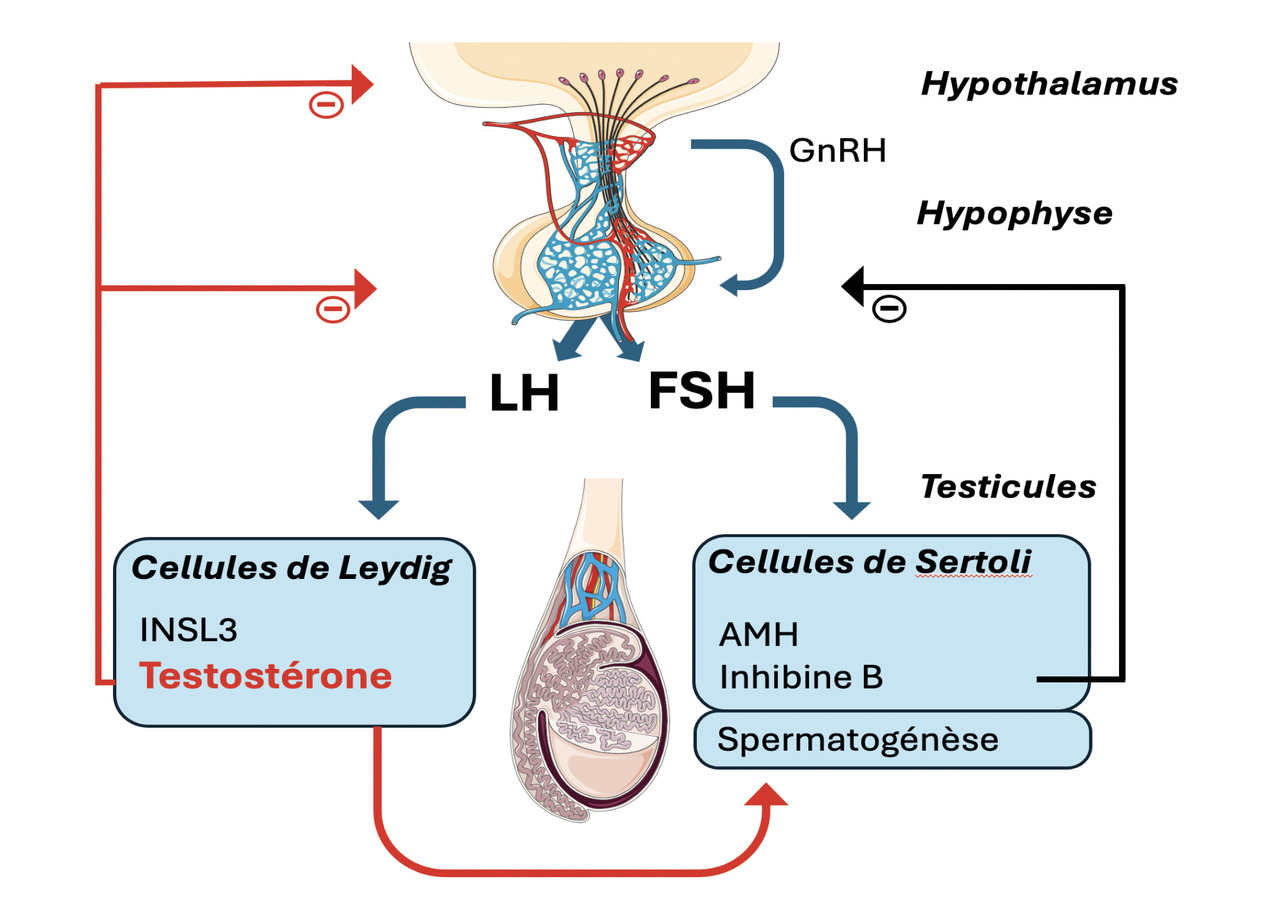


L’hypogonadisme masculin concernerait environ 1/200 hommes adultes, mais il est largement sous-diagnostiqué.
Il ne faut pas hésiter à poser des questions sur des troubles de la libido et des signes d’impuissance, éléments révélateurs d’un hypogonadisme.
La recherche de la cause centrale ou périphérique est indispensable avant de traiter.
Le dosage de la testostéronémie totale le matin est l’examen de première intention. Il s’accompagne de la mesure de la LH et de la FSH.
Le pronostic d’infertilité dépend surtout de la cause et de l’âge au diagnostic.