La crise du Levothyrox consécutive au remplacement du médicament par une nouvelle formule dans des conditions jugées a posteriori extrêmement critiquables par les pouvoirs publics eux-mêmes suscite de nombreuses interrogations, dont l’une concerne l’étude de bioéquivalence qui a permis la substitution d’une formule à l’autre. À la suite de la publication dans La Revue du Praticien de l’article de Catherine Hill et Martin Schlumberger « Les deux formules de Levothyrox ne sont pas bioéquivalentes » (Rev Prat 2019;69:559-601), nous avons reçu deux réponses que nous publions sous le titre « Les deux formules de Levothyrox sont-elles bioéquivalentes ? » (1) et (2) suivies de celle des auteurs (3).
La rédaction
Nous remercions Coste, Bertagna, Zureik et Nicolas d’une part et Ramez d’autre part de nous donner l’occasion de préciser un certain nombre de points ; nos réponses sont résumées dans le tableau ci-contre.
Bioéquivalence moyenne ou bioéquivalence individuelle
Pour conclure à la bioéquivalence entre les deux formulations du Levothyrox, suffit-il de démontrer qu’elles ont le même effet moyen, ce qui correspond à la réglementation actuelle ? Nous pensons au contraire que les patients ont le droit de savoir quelle proportion des sujets ont des réactions très différentes quand ils ingèrent les deux formulations. Cette information est d’autant plus essentielle que la lévothyroxine est un médicament à marge thérapeutique étroite : chaque sujet a une dose de confort qui est variable d’un sujet à l’autre. Il faut donc étudier la dispersion des différences de biodisponibilité entre les deux formulations (bioéquivalence individuelle) et pas seulement la moyenne de ces différences (bioéquivalence moyenne). Pour illustrer ce point, on pourrait imaginer trois sujets recevant chacun successivement les deux formulations : si les différences entre les deux formulations étaient de -30 %, 0 % et 30 %, la moyenne des différences (évaluée par l’étude de la bioéquivalence moyenne) serait nulle, mais un sujet serait en sous-dosage et un autre sujet en surdosage (c’est ce que montre l’étude de la bioéquivalence individuelle).
L’étude de Concordet et al., comme la nôtre, utilise donc les résultats de l’essai du laboratoire Merck pour étudier cette dispersion entre sujets des différences de réponses à l’ingestion des deux formulations. C’est une étude des mêmes données en fonction d’un objectif différent qui correspond à la situation en France, où une substitution a été imposée à des millions de patients.
Limite de l’étude de la bioéquivalence moyenne : son résultat dépend du nombre de sujets étudiés
Les autorités demandent aux laboratoires d’établir la bioéquivalence moyenne en se fondant sur l’intervalle de confiance du rapport entre les deux biodisponibilités moyennes qui doit être entièrement entre 0,90 et 1,11. Si le rapport est assez proche de 1 (il est égal à 0,99 dans l’analyse de Merck), il suffit d’inclure suffisamment de sujets pour obtenir un intervalle de confiance dans la fourchette de 0,90 à 1,11 exigée a priori par les autorités. En effet, la largeur de l’intervalle de confiance (la fourchette de précision) d’une moyenne diminue quand le nombre des sujets étudiés augmente : la précision d’une estimation augmente avec la taille de la population étudiée.
Ainsi plus on augmente le nombre de sujets, plus on a de chances de démontrer la bioéquivalence en moyenne. Ce résultat contre-intuitif a perturbé certains statisticiens qui se sont appuyés sur le principe de base : plus le nombre de sujets est grand plus le résultat est fiable, et n’ont pas vu qu’une moyenne entre 0,90 et 1,11 pourrait toujours avoir un intervalle de confiance inclus dans l’intervalle cible 0,90-1,11 si on augmentait le nombre de sujets. Les essais de bioéquivalence incluent en général entre 12 et 36 sujets, alors que l’essai de Merck en a inclus 204.
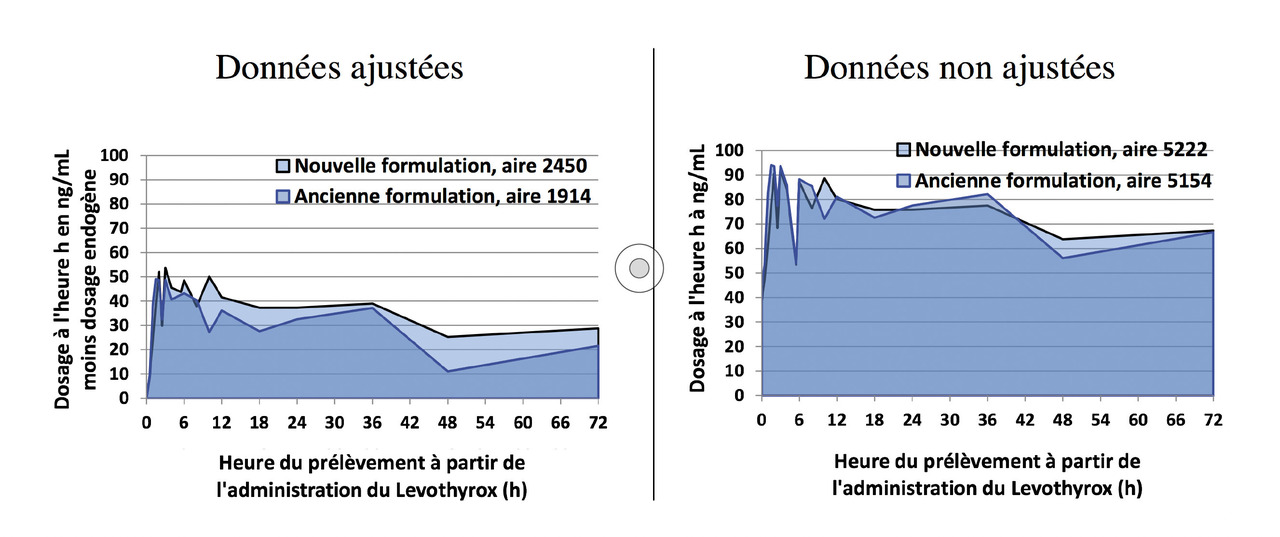
Soustraction ou non de la valeur basale de la lévothyroxine, analyse ajustée ou non ajustée
Coste et al. contestent la nécessité de soustraire la valeur de la lévothyroxine endogène observée chez chaque sujet avant l’administration du médicament (moyenne des trois valeurs observées une demi-heure avant, un quart d’heure avant et au moment de la prise du médicament) pour étudier la biodisponibilité des deux formulations de lévothyroxine exogène administrées à H0, l’étude allant jusqu’à H72. La
Interprétation de l’analyse individuelle ajustée
L’analyse que nous avons proposée ne fait que décrire la variation entre sujets de la biodisponibilité des deux formulations de lévothyrox. Nous ne disons pas quelle variabilité est acceptable. S’il faut définir une marge acceptable, celle-ci n’a pas de raison d’être identique pour la bioéquivalence moyenne et pour la bioéquivalence individuelle.
Autre limite de l’étude de Merck et de son analyse
Le Dr Ramez a raison, avec les données dont on dispose pour comparer les deux formulations de Levothyrox, c’est-à-dire avec une seule mesure par formulation pour chaque sujet, si on observe une différence entre les deux formules chez un même sujet, on ne sait pas quelle différence aurait été observée si on avait simplement refait la mesure une seconde fois avec la même formule chez le même sujet. L’étude de Merck ne répond qu’aux exigences des autorités française et européenne mais ne répond pas aux exigences de l’autorité des États-Unis, la Food and Drug Administration (FDA). En Europe, il suffit que chaque sujet prenne une fois l’ancienne formulation (A) et une fois la nouvelle formulation (N) dans un ordre aléatoire (dans l’étude 102 sujets ont reçu A puis N et 102 ont reçu N puis A). Aux États-Unis, la FDA demande que chaque sujet reçoive deux fois chaque formulation, la moitié des sujets recevant A puis N, et ensuite A puis N et l’autre moitié recevant N puis A, et ensuite N puis A. Cela permet d’étudier la variabilité intrinsèque de la biodisponibilité pour chaque sujet, c’est-à-dire la variabilité de l’aire sous la courbe entre les deux mesures du même sujet avec la même formulation.
Il est donc exact qu’avec les données de l’étude de Merck, on ne peut pas dissocier la variabilité intra-sujet entre formulations qui nous intéresse de la variabilité intra-sujet intra-formulations qui ne peut pas être évaluée. Cependant, si la réponse individuelle des patients à une même formulation était très variable, équilibrer leur traitement serait très difficile, même sans changer de médicament.
Variation entre formulations de la biodisponibilité individuelle et symptômes observés en France
Nous n’avons jamais prétendu que le changement de formulation pourrait expliquer tous les symptômes rapportés. Comme Concordet et al., nous pensons que les variations de bioéquivalence individuelle observées peuvent expliquer une partie des problèmes. En tout état de cause, l’étude de Merck ne permet pas d’exclure la responsabilité de la nouvelle formulation dans la survenue des signes et symptômes dont se sont plaints les sujets traités par lévothyroxine.
Analyse sur 10 sujets
Notre analyse a bien porté sur les 204 sujets de l’étude et non sur 10 sujets, comme le suggèrent Coste et al. Les données de 10 sujets ont été présentées pour illustrer schématiquement les données et les calculs.
Conclusion
Ce qui peut apaiser les conflits c’est d’écouter les patients et d’essayer de comprendre pourquoi un si grand nombre d’entre eux ont été mis en difficulté par le changement de formulation. Nous persistons à dire que la variabilité intra-sujet de la biodisponibilité des deux formulations de lévothyroxine est susceptible d’expliquer une partie des symptômes observés.
Il semble raisonnable d’envisager de changer la façon d’étudier les différentes formulations de lévothyroxine, au minimum en répliquant l’administration de chaque formulation selon les recommandations de la FDA, et en analysant les données en tenant compte du facteur sujet. V
Dans cet article
- Bioéquivalence moyenne ou bioéquivalence individuelle
- Limite de l’étude de la bioéquivalence moyenne : son résultat dépend du nombre de sujets étudiés
- Soustraction ou non de la valeur basale de la lévothyroxine, analyse ajustée ou non ajustée
- Interprétation de l’analyse individuelle ajustée
- Autre limite de l’étude de Merck et de son analyse
- Variation entre formulations de la biodisponibilité individuelle et symptômes observés en France
- Analyse sur 10 sujets
- Conclusion