La leucémie lymphoïde chronique (LLC) est une hémopathie lymphoïde chronique liée à l’envahissement progressif du sang, de la moelle osseuse et des organes lymphoïdes secondaires par des petits lymphocytes B monoclonaux, d’aspect mature et présentant un phénotype caractéristique. La lymphocytose clonale est, par définition, supérieure à 5 000 éléments/mm3 (5 G/L) et persistante dans le temps.
Épidémiologie (rang A)
La LLC a une incidence de 2 à 4 cas/100 000 habitants/an. Sa fréquence augmente avec l’âge : très rare avant 40 ans, l’âge médian au diagnostic est de 72 ans. Comme pour un certain nombre d’hémopathies, le diagnostic est plus souvent porté chez les hommes (environ 55 % des cas) que chez les femmes (environ 45 % des cas).
Étiologie (rang C)
La cause de la LLC reste inconnue à ce jour. Si les facteurs environnementaux classiques (toxiques, benzène ou irradiations) ne semblent pas jouer un rôle important dans la physiopathogénie, le rôle d’une stimulation antigénique initiale et/ou persistante du clone tumoral est supposé, notamment du fait de l’existence d’une sur-représentation de certains variants de la région génique codant pour la partie variable des immunoglobulines.
Les formes familiales de LLC sont rares : il ne s’agit donc pas d’une maladie héréditaire. Cependant, il existe un sur-risque de développer une hémopathie lymphoïde B de type LLC, maladie de Waldenström ou leucémie à tricholeucocytes dans l’entourage proche d’un patient atteint de LLC, suggérant l’existence d’un terrain génétique favorisant.
Circonstances de découverte (rang A)
La découverte d’une LLC est le plus souvent fortuite, devant une lymphocytose sanguine chez un sujet asymptomatique, sur un hémogramme demandé à titre systématique (plus de la moitié des cas).
Parfois, c’est la découverte d’un syndrome tumoral (adénopathies superficielles et/ou splénomégalie) ou, plus rarement, une complication auto-immune ou infectieuse qui conduisent à la réalisation de l’hémogramme.
L’étude des hémogrammes antérieurs est primordiale et permet de juger du caractère chronique de la lymphocytose.
Diagnostic positif (rang A)
Présentation clinique classique au diagnostic
Des adénopathies superficielles sont retrouvées par un examen clinique minutieux dans environ 70 % des cas. Elles sont classiquement de taille modérée, bilatérales, symétriques, indolores et mobiles par rapport aux plans superficiels et profonds. Une splénomégalie est observée dans environ 20 % des cas ; la présence d’une hépatomégalie est plus rare.
L’atteinte d’autres organes, comme la peau, les amygdales, le tube digestif, la plèvre, les poumons, les reins ou encore le système nerveux central, est décrite mais rarement rencontrée.
Analyse des lymphocytes
La clé du diagnostic est l’étude du sang périphérique.
Hémogramme et frottis sanguin
L’hémogramme est associé systématiquement à une étude morphologique des lymphocytes sur le frottis sanguin (coloration de May-Grünwald Giemsa).
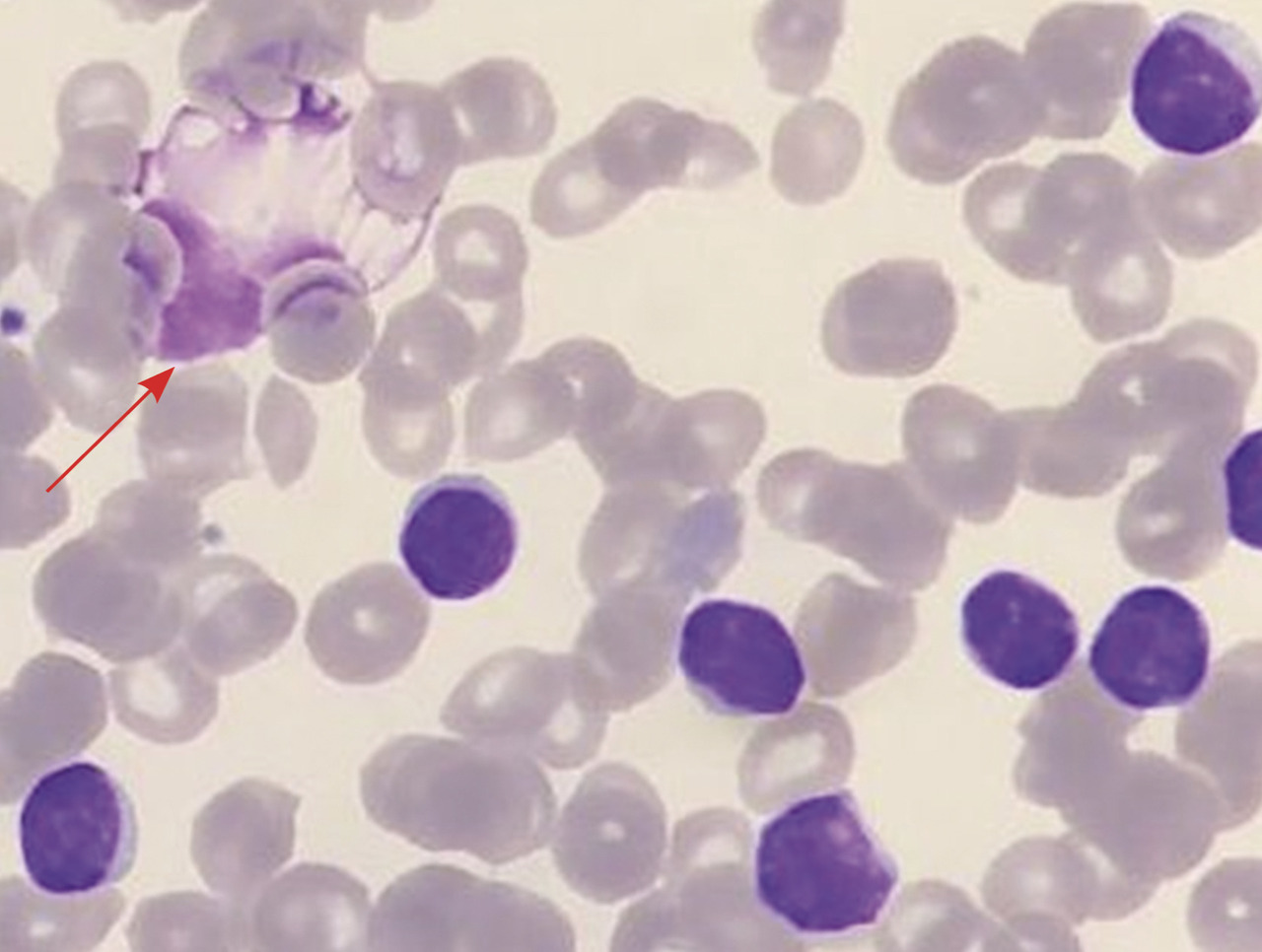
Une lymphocytose supérieure à 5 G/L est constante et indispensable pour poser le diagnostic de LLC. Cette lymphocytose atteint un niveau variable d’un patient à l’autre, pouvant dépasser le seuil de 200 G/L. Les cellules sont d’aspect classiquement monomorphe et très proche de celui des lymphocytes matures normaux (taille petite, cytoplasme peu abondant, sans signe d’activation, chromatine dense et mottée). Le frottis sanguin révèle fréquemment la présence de cellules altérées et de noyaux nus, appelés « ombres de Gumprecht » (figure).
Une anémie est présente dans 30 % des cas environ et se révèle significative (inférieure à 10 g/dL) dans 10 % des cas. Le compte des réticulocytes et la recherche de signes d’hémolyse sont indispensables pour en expliquer le mécanisme qui, en lien avec la LLC, peut se révéler multiple :
- insuffisance médullaire (réticulocytes abaissés, myélogramme montrant une infiltration tumorale par des cellules identiques aux cellules lymphocytaires circulantes) ;
- hémolyse auto-immune (haptoglobine effondrée, bilirubine libre élevée, réticulocytes augmentés, lactate déshydrogénase [LDH] augmentée, test direct à l’antiglobuline [test de Coombs direct] positif) ;
- hypersplénisme (en cas de volumineuse splénomégalie) ;
- érythroblastopénie auto-immune (réticulocytes inférieurs à 10 G/L, myélogramme montrant une raréfaction des érythroblastes et/ou un blocage de maturation de la lignée érythrocytaire).
Attention ! ne pas attribuer de facto une anémie à la LLC, notamment en cours de suivi à long terme et évoquer un mécanisme indépendant (comme une carence en fer).
Une thrombopénie est retrouvée dans 15 % des cas, significative (inférieure à 100 G/L) dans 10 % des cas. Sa cause est souvent multifactorielle et difficile à préciser (insuffisance médullaire, auto-immunité, hypersplénisme, etc.).
Une neutropénie est inconstante. Le chiffre réel des polynucléaires neutrophiles est parfois difficile à apprécier, notamment en cas de grande lymphocytose.
Immunophénotypage des lymphocytes sanguins (rang B)
Réalisé par cytométrie en flux, l'immunophénotypage est indispensable pour porter le diagnostic de LLC. Il caractérise le phénotype anormal des cellules lymphoïdes tumorales appartenant au compartiment B (CD19, CD20), monotypiques (expression d’une seule chaîne légère kappa ou lambda) et remplissant quatre ou cinq des critères du score dit de Matutes ou du RMH (Royal Marsden Hospital) [pour information : CD5 (+) – marqueur habituel du compartiment lymphocytaire T –, faible densité des immunoglobulines de surface, CD23 (+), CD79b (-), FMC7 (-)]. La présence d’un score de Matutes supérieur ou égal à 4 définit le diagnostic de LLC typique, tandis que des scores de 0, 1 ou 2 sont plus en faveur de lymphocytoses d’autre nature. Un score égal à 3 est parfois concordant avec un diagnostic de LLC dite « atypique ».
Bilan sérique complémentaire indispensable
L’électrophorèse des protéines sériques est systématique. Elle peut être normale, notamment au diagnostic. Une hypogammaglobulinémie est fréquente au cours de l’évolution de la maladie (60 % des patients). Un dosage pondéral des immunoglobulines peut alors compléter le bilan. La présence d’un pic monoclonal, le plus souvent modéré et de type IgM sur l’immunofixation sérique, est retrouvé dans environ 10 % dans cas.
Un test de Coombs direct est également systématique. Il est positif chez environ 5 % des patients, nécessitant la recherche d’une hémolyse associée (réticulocytes, bilirubine libre, LDH, haptoglobine), inconstante.
Bilan radiologique (rang C)
La taille des adénopathies profondes étant, dans la LLC, le plus souvent corrélée à celle des adénopathies superficielles, il n’est pas nécessaire d’effectuer un examen tomodensitométrique thoraco-abdomino-pelvien (TDM-TAP) au diagnostic. Cet examen est à faire en cas de suspicion de compression d’organe et/ou avant de débuter un traitement, spécialement dans les protocoles de recherche clinique.
La tomodensitométrie par émission de positons (TEP) n’est absolument pas pratiquée en routine dans la LLC. Cet examen n’est indiqué que lorsque l’on suspecte une transformation de la LLC en lymphome agressif (syndrome de Richter).
Autres examens complémentaires
Le myélogramme n’a pas d’intérêt dans le diagnostic de la LLC. Il montrerait une infiltration lymphoïde importante par des cellules identiques à celles du sang périphérique. Il peut être réalisé pour explorer le mécanisme d’une cytopénie, notamment le caractère central ou périphérique d’une thrombopénie.
La biopsie ostéomédullaire et la biopsie-exérèse ganglionnaire ne sont indiquées que dans les diagnostics difficiles et/ou en cas de suspicion de syndrome de Richter.
Formes cliniques voisines (rang A)
À côté de la LLC classique précédemment présentée, on décrit deux entités clinico-biologiques que l’on peut considérer comme des présentations différentes.
La lymphocytose B monoclonale est définie par l’existence d’un clone tumoral circulant de phénotype identique à la LLC mais dont la lymphocytose sanguine B monotypique est inférieure à 5 G/L. L’examen clinique et l’hémogramme sont par définition normaux dans ce cas. La présence d’une lymphocytose B monoclonale peut précéder l’apparition d’une authentique LLC, avec un taux d’évolution de 1 % par an. En l’absence d’évolution vers une LLC, les sujets avec une lymphocytose B monoclonale ont une survie identique à celle de la population générale.
Le lymphome lymphocytique est défini par l’existence d’un syndrome tumoral isolé, lié à un envahissement des organes lymphoïdes secondaires (ou plus rarement d’un autre organe) par des cellules tumorales monoclonales dont le phénotype est celui d’une LLC. Dans ce cas, on ne retrouve pas ou peu de cellules tumorales dans le sang (lymphocytose sanguine B monotypique inférieure à 5 G/L), et l’infiltration médullaire ne doit pas entraîner de cytopénies. Le diagnostic est alors porté le plus souvent sur une analyse histologique.
La leucémie prolymphocytaire B (rang C) est une autre entité pouvant être considérée comme une évolution ou un diagnostic différentiel de la LLC. Dans la LLC, un faible pourcentage de cellules peut avoir un aspect prolymphocytaire. Un taux supérieur à 15 % est défavorable. Lorsque ce taux est supérieur à 55 %, il peut s’agir d’une transformation prolymphocytaire ou d’une leucémie prolymphocytaire B de novo. L’évolution de ces formes est agressive.
Diagnostics différentiels (rang C)
Le diagnostic de LLC ne pose habituellement pas de problème de diagnostic différentiel, notamment depuis que l’immunophénotypage lymphocytaire est disponible en routine.
Les lymphocytoses polyclonales aiguës, essentiellement réactionnelles à des infections virales, sont facilement éliminées sur le contexte clinique totalement différent (sujet jeune, symptomatologie bruyante), l’aspect cytologique des lymphocytes sur le frottis (aspect de lymphocytes activés, voire syndrome mononucléosique) et l’historique de la lymphocytose d’évolution aiguë.
En l’absence d’arguments pour une lymphocytose polyclonale aiguë, l’analyse cytologique du frottis sanguin et l’immunophénotypage des lymphocytes circulants sont les examens clés permettant d’orienter vers d’autres lymphopathies monoclonales de forme leucémisée (c’est-à-dire avec passage de cellules tumorales dans le compartiment sanguin).
Les lymphomes non hodgkiniens de phénotype B avec passage sanguin de cellules lymphomateuses constituent un autre diagnostic différentiel. Les plus fréquents sont des lymphomes indolents : le lymphome folliculaire, le lymphome à cellules du manteau, le lymphome de la zone marginale avec ou sans lymphocytes villeux, la macroglobulinémie de Waldenström (lymphome lymphoplasmocytaire médullaire avec immunoglobuline monoclonale de type M) ou encore la leucémie à tricholeucocytes. L’aspect cytologique des cellules lymphomateuses au frottis sanguin est différent et plus atypique. L’analyse des lymphocytes du sang par l’immunophénotypage (phénotype B et score de Matutes le plus souvent inférieur à 2) et le caryotype peuvent apporter une forte orientation. Mais la biopsie médullaire ou ganglionnaire est généralement requise pour confirmer le diagnostic.
Les lymphomes de phénotype T ou NK sont aussi à éliminer tels que la leucémie à grands lymphocytes granuleux, la leucémie prolymphocytaire T ou encore le syndrome de Sézary, qui est associé à une atteinte cutanée. Dans certaines populations à risque, du fait de l’origine géographique (Caraïbes, Iran, Japon…), doit être évoquée la possibilité d’une leucémie/lymphome T de l’adulte associé au virus HTLV- 1, dont il peut exister des formes chroniques et indolentes. Une analyse globale (morphologique, phénotypique et génétique) permet le diagnostic de ces entités. Le score de Matutes, décrit pour les hémopathies B, ne s’applique pas à ces lymphomes T.
Les lymphocytoses polyclonales chroniques, généralement modérées (4 - 6 G/L) sont facilement exclues par le contexte et l’immunophénotypage lymphocytaire : hypo- ou asplénisme ; lymphocytose chronique à lymphocytes binucléés des sujets fumeurs, prédominante chez les sujets de sexe féminin ; lymphocytose induite par le natalizumab (traitement de la sclérose en plaques).
Pronostic (rang A)
Le pronostic de la LLC a longtemps été considéré comme imprévisible : alors que certains patients décédaient dans les mois suivant le diagnostic, la survie d’autres patients était prolongée et non différente de celle de la population générale.
Classifications de pronostic à critères clinico-biologiques
D’anciennes études de grandes séries de patients ont permis de mettre au point des classifications pronostiques simples dont deux principales : celle dite « Rai », en cinq stades, utilisée aux Etats-Unis, et celle de Binet, en trois stades, largement utilisée en Europe (tableau).
Les aires ganglionnaires lymphoïdes, définies pour l’évaluation de ces classifications, sont au nombre de cinq : aires cervicale, axillaire et inguinale, foie, rate. L’atteinte de ces aires doit être déterminée par l’examen physique du patient. La tomodensitométrie n’intervient pas dans cette évaluation.
L’anémie et la thrombopénie ont une valeur pronostique considérable et apparaissent comme un signe de gravité majeur dans les deux classifications.
Il faut cependant prendre en considération que ces classifications ont été créées avant l’avènement des thérapies ciblées, thérapeutiques qui ont révolutionné la prise en charge des LLC et amélioré le pronostic. Actuellement, elles restent utilisées pour l’indication thérapeutique, alors que d’autres marqueurs pronostiques (statut IgVH, statut TP53) sont pris en compte pour le choix de la thérapeutique.
Autres paramètres biologiques requis (rang C)
Des anomalies cytogénétiques sont retrouvées dans 50 à 80 % des cas, en utilisant le caryotype et la fluorescence par hybridation in situ (FISH) en complément du caryotype conventionnel réalisé sur les cellules tumorales. À ces anomalies s’ajoutent des mutations acquises, aujourd’hui détectées en biologie moléculaire par la technique de NGS (Next Generation Sequencing). Certaines ont un impact pronostique majeur.
Altérations de TP53. La délétion du bras court du chromosome 17 (délétion 17p) emporte le locus du gène codant la protéine TP53, qui est un gardien du génome. Une mutation du gène TP53, alors non emporté par une délétion 17p, a la même valeur péjorative. Ces altérations de TP53 constituent le facteur pronostique le plus défavorable connu à ce jour de la LLC. Elles sont associées à une efficacité très réduite des chimiothérapies conventionnelles et ont un impact négatif variable sur les thérapeutiques ciblées.
La trisomie 12 est associée à un aspect cytologique atypique.
La délétion 11q emporte le gène ATM et confère un mauvais pronostic ; l’atteinte tumorale ganglionnaire est plus fréquente.
La délétion 13q est associée à un meilleur pronostic.
Les caryotypes complexes et ultracomplexes, se définissant comme plusieurs anomalies caryotypiques, sont de mauvais pronostic.
Le statut mutationnel du gène des chaînes lourdes des Ig (IgVH) reflète la « maturation » de la cellule initiatrice de LLC. Physiologiquement, le gène IgVH est muté dans le centre germinatif après la rencontre du lymphocyte B avec un antigène, dans le but d’augmenter l’affinité de l’immunoglobuline pour l’antigène : c’est le phénomène d’hypermutation somatique. Dans la LLC, le statut du gène IgVH a un impact pronostique démontré : les LLC immatures, sans mutation IgVH, ont un pronostic plus défavorable.
Parmi ces anomalies, certaines sont à rechercher impérativement car elles ont un fort impact sur le pronostic et la réponse thérapeutique :
- les altérations TP53 doivent être recherchées avant chaque ligne thérapeutique, car la fréquence de détection varie en augmentant avec le nombre de lignes ;
- le statut mutationnel IgVH, qui reste indéfiniment le même dans la LLC, doit être déterminé une seule fois dans la prise en charge, généralement au diagnostic ou avant la première ligne thérapeutique.
Autres paramètres décrits mais non requis en routine (rang C)
D’autres marqueurs biologiques simples sont décrits comme associés à un mauvais pronostic : taux sériques augmentés de LDH et de bêta- 2 -microglobuline.
Un temps de doublement de la lymphocytose inférieur à douze mois est également un facteur de mauvais pronostic.
Évolution
Histoire naturelle (rang C)
Du fait de l’augmentation des prescriptions d’hémogrammes systématiques, le diagnostic de LLC est posé beaucoup plus tôt qu’auparavant : la découverte est alors fortuite et intervient chez des sujets asymptomatiques dans 70 % des cas.
Au moins la moitié des patients diagnostiqués au stade A de la classification de Binet restent stables pendant très longtemps (parfois plus de dix ans) : leur survie est alors identique à celle de la population générale du même âge.
Chez les autres patients, la masse tumorale augmente progressivement, entraînant une majoration de la lymphocytose, des cytopénies et/ou du syndrome tumoral (ganglionnaire, splénique et/ou hépatique).
La LLC reste aujourd’hui encore incurable, en dehors du recours à l’allogreffe de cellules souches hématopoïétiques dont l’utilité a été considérablement réduite par le développement des thérapeutiques ciblées.
Schématiquement, il est classique de considérer, au diagnostic d’une LLC, que :
- un tiers des patients (principalement de stade A) auront une évolution indolente ne nécessitant jamais de traitement ;
- un tiers des patients auront une évolution indolente, nécessitant un traitement à moyen ou long terme (au-delà de cinq ans de suivi) ;
- un tiers des patients auront une évolution rapide, nécessitant un traitement à court terme (dans les cinq ans).
Ainsi, une proportion importante de patients ayant une présentation limitée à une lymphocytose sans cytopénie, sans adénopathie ni splénomégalie importante, ne recevront pas immédiatement de traitement. Ces patients devront être surveillés en consultation, en prenant soin de ne pas attribuer de facto tout nouvel événement à la LLC et en sachant qu’indépendamment de tout traitement, ils présentent une augmentation variable du risque infectieux. La décision de ne pas traiter une LLC doit être réfléchie, discutée et bien expliquée.
Traitement
(Rang B) Le traitement est fondé sur la présence de critères d’évolutivité décrits par l’International Workshop on Chronic Lymphocytic Leukaemia (IWCLL). Ces critères se retrouvent principalement chez des patients de stades B et C. Ces critères comprennent : cytopénies (anémie, thrombopénie, neutropénie), adénopathie ou splénomégalie volumineuse, altération de l’état général, cytopénie auto-immune, augmentation rapide de la lymphocytose.
(Rang C) Jusqu’alors, le traitement conventionnel était représenté par une association d’immunochimiothérapie. L’ère thérapeutique de la LLC a changé dans la dernière décennie, avec l’avènement des thérapies ciblées, dont les deux classes principales sont :
- les inhibiteurs au récepteur des cellules B (BCR) qui est suractivé et participe à la prolifération et l’expansion des lymphocytes B tumoraux. Ils comprennent essentiellement les inhibiteurs de la Bruton tyrosine kinase (BTK) dont ibrutinib, acalabrutinib, zanubrutinib, qui entraînent une inactivation de cette voie enzymatique, empêchant ainsi l’expansion du clone tumoral. Une autre classe existe, celle des inhibiteurs de la PI3 kinase (idélalisib), moins utilisée ;
- les inhibiteurs de BCL2 qui est une protéine antiapoptotique retrouvée à la surface des mitochondries du lymphocyte B. Elle régule l’apoptose et permet ainsi l’immortalité de la cellule tumorale dans la LLC. L’inhibiteur de BCL2 (vénétoclax) permet alors une réactivation des voies apoptotiques, entraînant la destruction des cellules tumorales.
Une immunothérapie est couramment associée au vénétoclax, il s’agit d’anticorps monoclonal anti-CD20 (rituximab ou obinutuzumab).
La chimiothérapie conventionnelle (fludarabine, bendamustine, cyclophosphamide, chlorambucil) a une place qui s’est considérablement réduite à des patients sélectionnés et non éligibles aux thérapies ciblées.
Le choix du traitement dépend de l’état général du patient, du spectre de ses comorbidités cliniques et biologiques, de son statut TP53 et du statut IgVH de ses lymphocytes tumoraux.
Complications
Les complications sont classiquement de trois ordres : infectieuses, auto-immunes et tumorales.
Complications infectieuses (rang B)
Le plus souvent observées dans les formes les plus avancées de la maladie. Elles relèvent de trois mécanismes, souvent associés.
Altération de l’immunité humorale innée et adaptative. L’hypogammaglobulinémie résulte de la prédominance du clone tumoral dans le compartiment B aux dépens des lymphocytes B constitutionnels entraînant une répression de synthèse. Elle s’aggrave classiquement avec l’administration des traitements, notamment les anticorps anti-CD20.
Altération de l’immunité cellulaire adaptative. Elle existe également avant traitement et se majore nettement après l’utilisation des thérapeutiques.
Altération de l’immunité innée. La neutropénie est rarement présente au diagnostic, mais sa fréquence et son implication dans les complications infectieuses augmentent nettement avec l’utilisation des thérapies ciblées.
Les infections sont la première cause de morbidité et de mortalité dans la LLC. Si les infections bactériennes à germes communautaires restent les plus fréquentes, les thérapeutiques utilisées ont modifié le spectre des infections, avec l’émergence d’infections à germes opportunistes comme la pneumocystose, la listériose ou la légionellose, et d’infections virales (herpès, zona), voire fungiques.
La pandémie liée au SARS-CoV- 2 a bien démontré une capacité réduite de réponse vaccinale (ARN) chez les patients atteints de LLC.
Les patients atteints de LLC sont donc à considérer comme des sujets (fortement) immunodéprimés, surtout pendant et après traitement :
- les vaccinations doivent être proposées dès le diagnostic et au plus tard avant traitement : antipneumococcique (conjugué), antigrippale, anti-SARS-CoV- 2 (ARN) et antivaricelle-zona (recombinant) ;
- des prophylaxies anti-infectieuses primaires (cotrimoxazole contre Pneumocystis, valaciclovir contre l’herpès simplex virus et le virus varicelle-zona) sont obligatoirement associées aux traitements spécifiques de la maladie ;
- une substitution en immunoglobulines polyvalentes est indiquée en cas de complications infectieuses sévères et/ou répétées ;
- enfin, il convient de garder à l’esprit ce statut d’immunodéprimé dans la gestion des épisodes infectieux d’allure banale, afin de ne pas méconnaître une évolution péjorative ou de retarder une indication d’antibiothérapie.
Complications auto-immunes (rang C)
Les complications auto-immunes sont caractéristiques, notamment les cytopénies auto-immunes.
L’anémie hémolytique auto-immune (AHAI) est la plus fréquente : le test direct à l’antiglobuline (Coombs direct érythrocytaire) est le plus souvent de type IgG (plus ou moins associé au complément) à anticorps chauds. Une anémie hémolytique auto-immune est retrouvée chez 7 à 10 % des patients atteints de leucémie lymphoïde chronique.
Les thrombopénies auto-immunes sont moins fréquentes (1 à 5 % des patients) et sont surtout difficiles à mettre en évidence.
Les érythroblastopénies auto-immunes sont rares et caractérisées par une anémie totalement arégénérative (réticulocytes < 10 G/L), avec disparition des précurseurs de la lignée érythrocytaire dans la moelle osseuse.
Complications tumorales (rang C)
Une évolution de la LLC en un lymphome agressif survient avec une fréquence encore mal connue (2 à 10 % des patients atteints de LLC), constituant le classique syndrome de Richter. Celui-ci résulte le plus souvent d’une transformation d’un sous-clone de LLC. Les signes cliniques et biologiques devant le faire suspecter sont ceux usuellement retrouvés au diagnostic de lymphome de haut grade : augmentation rapide de volume tumoral, altération de l’état général, sueurs nocturnes, fièvre et/ou perte de poids, s’associant à une élévation du taux des LDH, à une thrombopénie, à une hypercalcémie et parfois à un syndrome inflammatoire biologique. L’analyse histologique en fait le diagnostic positif. Classiquement, elle décrit un envahissement tumoral par de larges plages de grandes cellules B sur un fond de cellules de la LLC sous-jacentes, déstructurant l’architecture de l’organe étudié. Dans 95 % des cas, il s’agit d’un lymphome B diffus à grandes cellules. Le pronostic du syndrome de Richter est très sombre.
Enfin, il est classiquement admis que le risque de cancer est accru chez les patients atteints de LLC, sans prédominance d’organe. Il convient surtout d’insister sur la nécessité de bien organiser le suivi prolongé et potentiellement anxiogène de la LLC sans détourner l’attention des différents acteurs du soin des autres actions de prévention et de dépistage.
Conclusion
La leucémie lymphoïde chronique est l’hémopathie la plus fréquente. Souvent de découverte fortuite, son évolution est imprévisible et nécessite un suivi spécialisé prolongé. Le rôle des différents acteurs de la prise en charge médicale de ces patients est essentiel pour assurer la gestion des complications infectieuses et pour ne pas méconnaître les complications auto-immunes et tumorales. La gestion de l’anxiété, qui accompagne cette maladie tumorale chronique, et des comorbidités souvent associées à l’âge des patients sont également d’importance.
Message de l'auteur
La leucémie lymphoïde chronique peut constituer le sujet principal d’un dossier aux EDN.
Une première partie permettrait de vérifier les connaissances de base en hématologie, notamment sur les anomalies de l’hémogramme (hyperlymphocytose, mais aussi anémie ou thrombopénie) ou la conduite à tenir devant un tableau de poly-adénopathies. Une deuxième partie pourrait permettre de vérifier que le principe des examens de laboratoire, comme le frottis sanguin ou l’immunophénotypage, sont bien compris. Enfin, les dernières questions pourraient porter sur la conduite à tenir devant l’apparition de signes généraux évoquant une transformation en lymphome de haut grade, sur le diagnostic d’une anémie hémolytique auto-immune venant compliquer l’évolution ou encore sur la gestion des complications infectieuses spécifiques au terrain immunodéprimé.
Si le traitement de la leucémie lymphoïde chronique n’est pas au programme de cet item, les notions concernant la gestion des complications infectieuses (antibiothérapie, vaccinations, substitution en immunoglobulines polyvalentes) sont à l’intersection de plusieurs items et doivent donc être acquises.
La leucémie lymphoïde chronique (LLC) est une hémopathie lymphoïde d’évolution chronique dont la fréquence augmente avec l’âge.
Le diagnostic est posé sans examen invasif : l’hémogramme avec étude cytologique du frottis sanguin et l’immunophénotypage des lymphocytes circulants en cytométrie en flux sont les examens clés.
La classification de Binet reste un bon outil pronostique afin d’anticiper l’évolution la maladie. Les critères IWCLL d’évolutivité permettent de poser l’indication d’un traitement.
Il est indispensable de rechercher une altération de TP53 (délétion 17p et/ou mutation du gène TP53) avant chaque ligne de traitement, ainsi que le statut du gène IgVH une seule fois au cours de l’histoire de la LLC.
Devant les complications auto-immunes et tumorales, les complications infectieuses sont au premier plan. Elles doivent toujours être redoutées et bénéficier d'une prévention optimale.
Testez-vous sur cet item
Découvrez quelques quiz rédigés par les auteurs de cet item :
– Haute Autorité de santé
– Institut national du cancer
– Société française d’hématologie
– French Innovative Leukemia Organization (FILO)
 Encadrés
Encadrés