Une maladie à évoquer devant une cardiopathie hypertrophique ou une insuffisance rénale inexpliquée, des AVC précoces ou des douleurs des extrémités, aussi bien chez l’homme que chez la femme.
La maladie de Fabry est liée à des mutations du gène GLA, codant l’α-galactosidase A (αGalA). Comme de nombreuses enzymes lysosomales, elle participe au catabolisme de macromolécules, spécifiquement de glycosphingolipides, constituants essentiels des membranes plasmiques. Un défaut enzymatique entraîne l’accumulation de son substrat, le globotriaosylcéramide (Gb3) et de sa forme dé-acétylée soluble : globotriaosylsphingosine (lyso-Gb3). L’accumulation de Gb3 est lysosomale, intracellulaire mais aussi membranaire à l’origine de phénomènes inflammatoires : production de cytokines (tumor necrosis factor alpha [TNF-α], interleukines [IL]-1b, IL-6, IL-18), activation de l’immunité innée (toll-like receptor 4) et fibrose locale. Les tissus cibles sont les tissus riches en Gb3 : le système nerveux périphérique et central, le système cardiovasculaire dans son ensemble et les reins.
Épidémiologie
Au décours de campagnes de dépistage systématique chez des nouveaux nés asymptomatiques, on estime aujourd’hui la prévalence de la maladie à 1/10 000 naissances vivantes avec une surreprésentation du phénotype cardiaque par rapport au phénotype historique avec un ratio de 7:1. Du fait de la méconnaissance de la maladie, seuls 800 patients sont actuellement diagnostiqués en France. L’espérance de vie avec maladie de Fabry était estimée en 2009 à 58 ans chez les hommes et 75 ans chez les femmes, l’atteinte cardiaque étant la principale cause de décès. Cependant, les formes classiques (v. infra) étaient vraisemblablement surreprésentées à l’époque.
Transmission, génétique
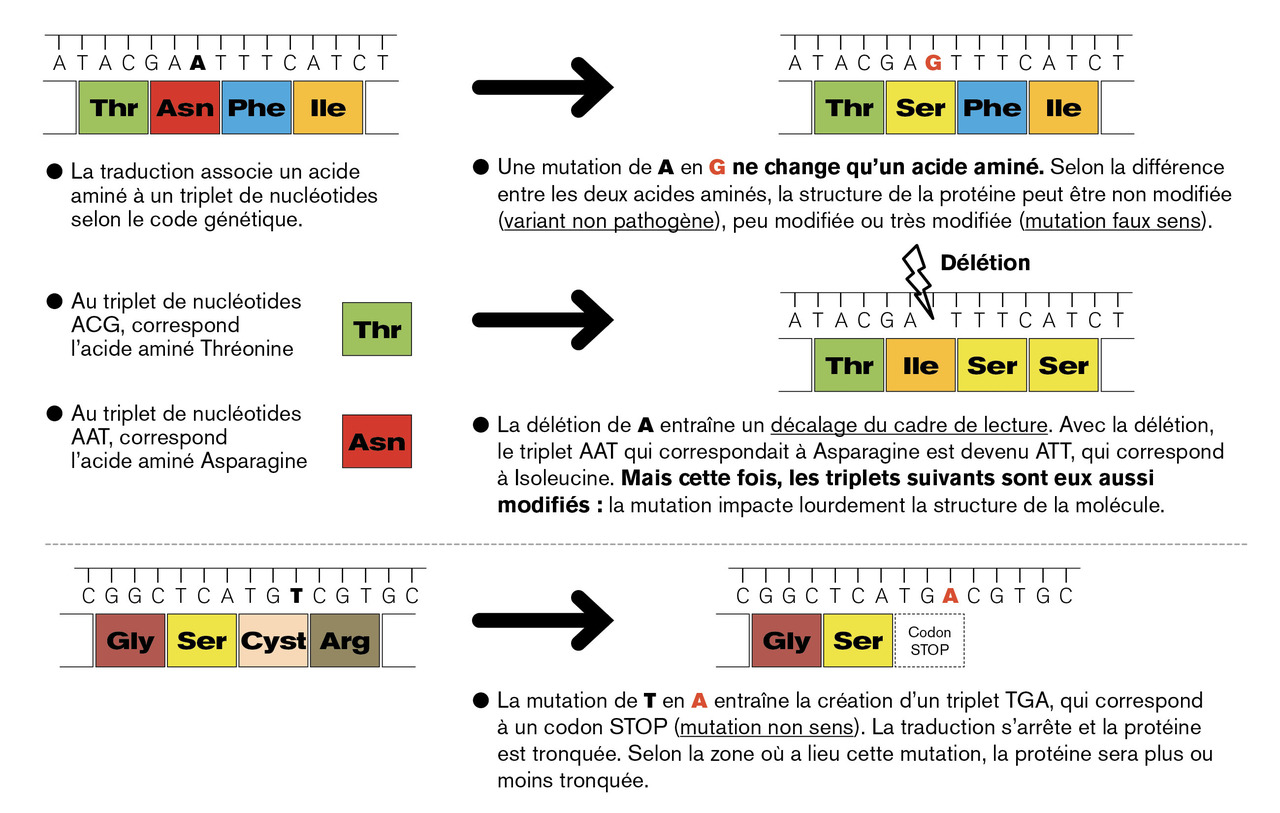
Le gène GLA est sur le chromosome X. Un homme porteur d’un X « atteint » est nécessairement malade. L’expression clinique dépend de la mutation dans la séquence d’ADN du gène (plus de 1 000 variants décrits) :
– la structure enzymatique n’est pas modifiée et l’enzyme fonctionnelle : la production d’αGalA est maintenue, il s’agit d’un variant non pathogène : pas de maladie ;
– la mutation affecte une structure clé, voire empêche totalement la production d’enzyme : l’activité résiduelle est nulle (cas de la plupart des délétions, décalages du site de lecture ou de certaines mutations faux-sens) : la maladie est sévère ;
– la mutation entraîne des modifications mineures à l’origine d’une enzyme instable ou moins efficace (mutations faux-sens) : l’activité enzymatique résiduelle est diminuée mais persiste, le phénotype est intermédiaire (fig. 1 ).
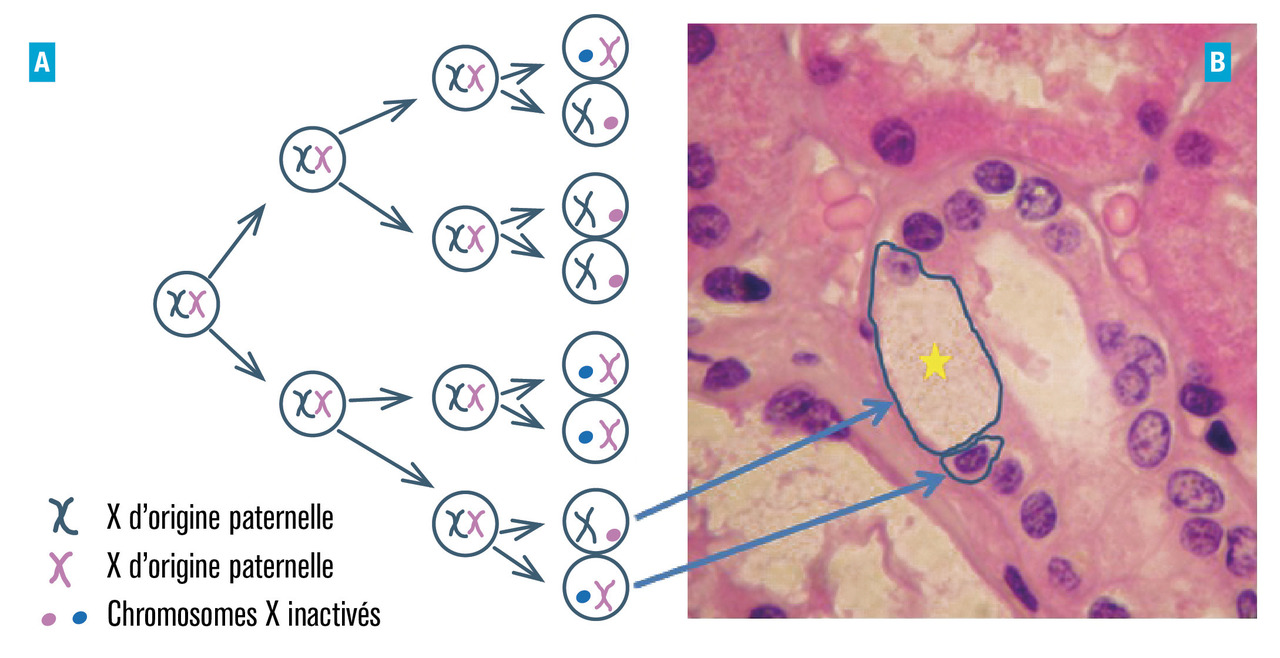
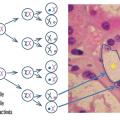
On a longtemps cru que les femmes porteuses d’un X sain n’étaient que vectrices. On sait aujourd’hui qu’elles peuvent être atteintes aussi sévèrement que les hommes du fait du phénomène de lyonisation (inactivation aléatoire d’un des 2 chromosomes X au stade embryonnaire dans chaque cellule) : au sein d’un même organe deux cellules voisines peuvent exprimer une enzyme fonctionnelle et une non-fonctionnelle (fig. 2 ) respectivement.
– la structure enzymatique n’est pas modifiée et l’enzyme fonctionnelle : la production d’αGalA est maintenue, il s’agit d’un variant non pathogène : pas de maladie ;
– la mutation affecte une structure clé, voire empêche totalement la production d’enzyme : l’activité résiduelle est nulle (cas de la plupart des délétions, décalages du site de lecture ou de certaines mutations faux-sens) : la maladie est sévère ;
– la mutation entraîne des modifications mineures à l’origine d’une enzyme instable ou moins efficace (mutations faux-sens) : l’activité enzymatique résiduelle est diminuée mais persiste, le phénotype est intermédiaire (
On a longtemps cru que les femmes porteuses d’un X sain n’étaient que vectrices. On sait aujourd’hui qu’elles peuvent être atteintes aussi sévèrement que les hommes du fait du phénomène de lyonisation (inactivation aléatoire d’un des 2 chromosomes X au stade embryonnaire dans chaque cellule) : au sein d’un même organe deux cellules voisines peuvent exprimer une enzyme fonctionnelle et une non-fonctionnelle (
Atteintes cliniques
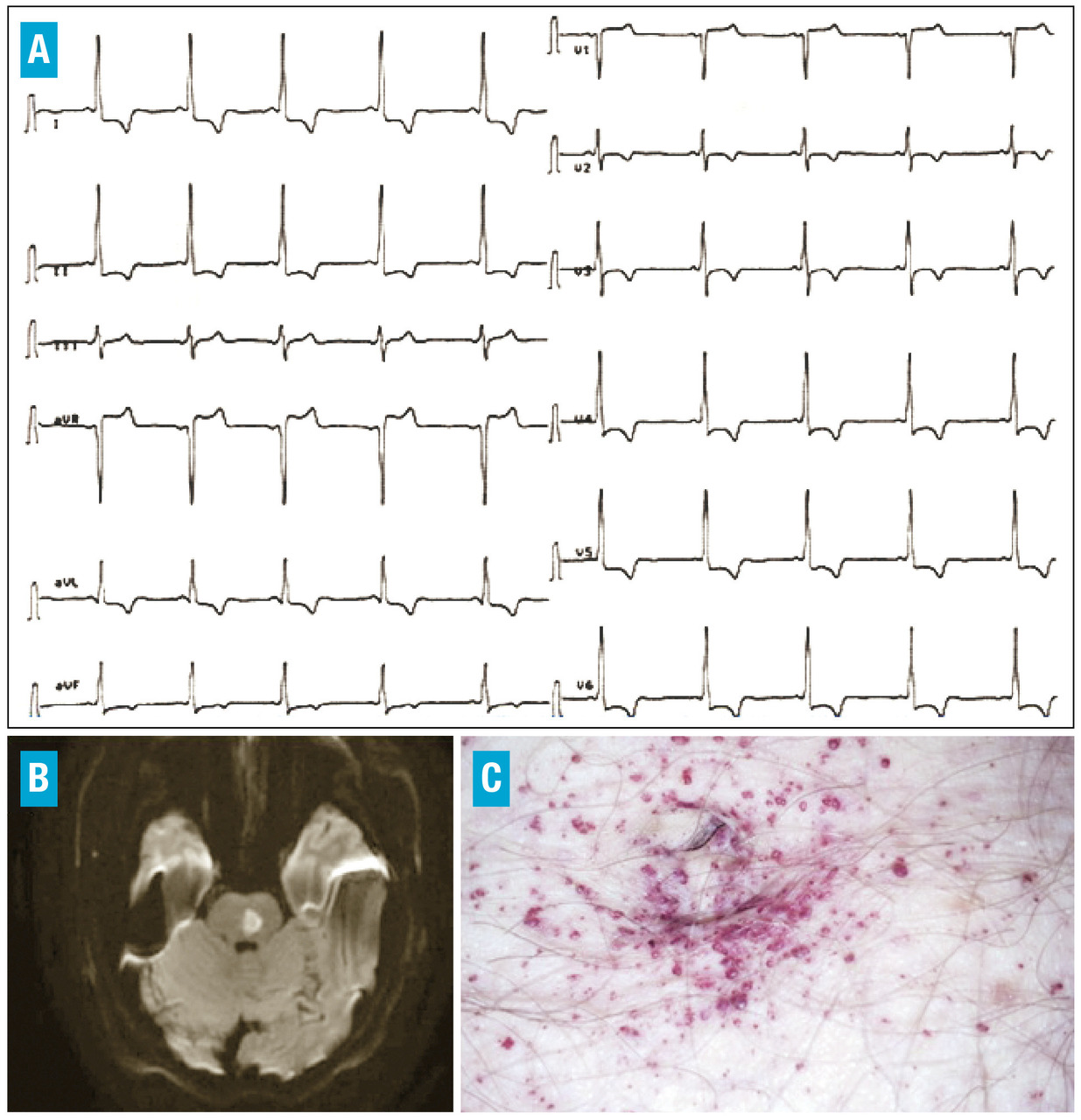
Elles sont présentées dans le tableau ci-contre et la figure 3 .1, 2
On décrit aujourd’hui deux formes cliniques : la forme classique et le variant cardiaque.
Dans la forme classique, toutes les atteintes sont observées. C’est la forme décrite initialement par Fabry et Anderson en 1898 caractérisée par trois phases évolutives :
– une phase précoce entre 0 et 20 ans, où prédominent les acroparesthésies, volontiers accompagnées d’intolérance au sport (hypohidrose) et d’asthénie. La présence d’une cornée verticillée et d’angiokératomes aide au diagnostic ;
– une phase quiescente, « lune de miel entre » 20 et 30 ans où on peut observer une amélioration spontanée des acroparesthésies alors que les dépôts s’accumulent, sans aucun symptôme ;
– une phase tardive, après 30 ans, où les atteintes d’organes deviennent symptomatiques : accidents vasculaires cérébraux, insuffisance rénale progressive, cardiopathie.
Dans la forme non classique ou « variant cardiaque », l’atteinte cardiaque est quasi exclusive (atteinte oto-rhino-laryngée possible). C’est de loin la forme la plus fréquente. On doit donc évoquer la maladie de Fabry devant une hypertrophie cardiaque sans obstacle à l’éjection ventriculaire gauche évidente (hypertension artérielle, rétrécissement aortique, bourrelet obstructif).
On décrit aujourd’hui deux formes cliniques : la forme classique et le variant cardiaque.
Dans la forme classique, toutes les atteintes sont observées. C’est la forme décrite initialement par Fabry et Anderson en 1898 caractérisée par trois phases évolutives :
– une phase précoce entre 0 et 20 ans, où prédominent les acroparesthésies, volontiers accompagnées d’intolérance au sport (hypohidrose) et d’asthénie. La présence d’une cornée verticillée et d’angiokératomes aide au diagnostic ;
– une phase quiescente, « lune de miel entre » 20 et 30 ans où on peut observer une amélioration spontanée des acroparesthésies alors que les dépôts s’accumulent, sans aucun symptôme ;
– une phase tardive, après 30 ans, où les atteintes d’organes deviennent symptomatiques : accidents vasculaires cérébraux, insuffisance rénale progressive, cardiopathie.
Dans la forme non classique ou « variant cardiaque », l’atteinte cardiaque est quasi exclusive (atteinte oto-rhino-laryngée possible). C’est de loin la forme la plus fréquente. On doit donc évoquer la maladie de Fabry devant une hypertrophie cardiaque sans obstacle à l’éjection ventriculaire gauche évidente (hypertension artérielle, rétrécissement aortique, bourrelet obstructif).
Circonstances et démarche diagnostiques
En cas d’atteinte clinique évocatrice,1 le diagnostic est fait chez l’homme par le dosage de l’activité de l’α-Gal A intraleucocytaire qui est abaissé, confirmé par le génotypage du gène GLA. Chez la femme, le diagonstic se fait par génotypage du gène GLA en première intention (l’activité de l’α-GalA intraleucocytaire peut être normale en cas d’inactivation intraleucocytaire de l’X atteint sans être le reflet de l’activité enzymatique existant dans les autres tissus).
Dans le cadre d’un dépistage familial, lorsqu’un cas index est découvert, on doit systématiquement proposer une enquête familiale. En pratique, pour les enfants et adolescents asymptomatiques, on propose le dépistage par recherche du variant GLA familial en fin d’adolescence.
Dans le cadre d’un dépistage familial, lorsqu’un cas index est découvert, on doit systématiquement proposer une enquête familiale. En pratique, pour les enfants et adolescents asymptomatiques, on propose le dépistage par recherche du variant GLA familial en fin d’adolescence.
Traitement
Le traitement repose sur trois axes : la prévention, le traitement symptomatique et la correction du déficit enzymatique.
Prévention et lutte contre les co-pathogènes
La prévention des atteintes d’organe est d’autant plus efficace que le diagnostic est précoce. On propose un programme de surveillance annuel, voire semestriel en fonction de la sévérité des atteintes. La prévention des risques cardiovasculaires est particulièrement importante car les organes cibles de la maladie de Fabry sont aussi ceux de l’athérome et du diabète.
Traitement symptomatique
Qu’il s’agisse de la prise en charge des acroparesthésies, des douleurs abdominales, de l’atteinte rénale, cardio- ou cérébrovasculaire, la prise en charge symptomatique ne diffère pas de celle des patients non atteints de maladie de Fabry. Les seules restrictions concernent l’usage de l’amiodarone et de la chloroquine susceptibles d’atténuer l’efficacité d’une enzymothérapie substitutive par modification du pH lysosomal.
Les douleurs neuropathiques sont traitées par antalgiques adaptés (gabapentine, prégabaline, antidépresseurs tricycliques, inhibiteurs de recapture de la sérotonine). Les angiokératomes peuvent être cautérisés au laser. Une néphroprotection est proposée par des inhibiteurs de l’enzyme de conversion ou des antagonistes AT1 de l’angiotensine II. La prise en charge de la cardiopathie fait souvent appel aux antiagrégants, anticoagulants et anti-arythmiques selon les indications cardiologiques symptomatiques. Le recours au stimulateur cardiaque est fréquent.
Le dépistage et la prise en charge spécialisée des troubles psychologiques et psychiatriques est primordiale dans ce cadre de maladie génétique, chronique et douloureuse où les troubles anxiodépressifs et les tentatives de suicides sont fréquents. Une orientation vers une association de patients est souhaitable.*
Les douleurs neuropathiques sont traitées par antalgiques adaptés (gabapentine, prégabaline, antidépresseurs tricycliques, inhibiteurs de recapture de la sérotonine). Les angiokératomes peuvent être cautérisés au laser. Une néphroprotection est proposée par des inhibiteurs de l’enzyme de conversion ou des antagonistes AT1 de l’angiotensine II. La prise en charge de la cardiopathie fait souvent appel aux antiagrégants, anticoagulants et anti-arythmiques selon les indications cardiologiques symptomatiques. Le recours au stimulateur cardiaque est fréquent.
Le dépistage et la prise en charge spécialisée des troubles psychologiques et psychiatriques est primordiale dans ce cadre de maladie génétique, chronique et douloureuse où les troubles anxiodépressifs et les tentatives de suicides sont fréquents. Une orientation vers une association de patients est souhaitable.*
Correction du déficit enzymatique
Enzymothérapie substitutive. Depuis 2001, deux enzymothérapies substitutives en perfusion intraveineuse disposent d’une autorisation de mise sur le marché :4, 5 l’ agalsidase alfa (Replagal : 0,2 mg/kg/14 j) et l’agalsidase bêta (Fabrazyme : 1 mg/kg/14 j). L’enzymothérapie atténue les douleurs, prévient le déclin de la fonction rénale et diminue l’épaississement myocardique à condition d’être débutée avant le développement de la fibrose, irréversible. De nombreux experts et une méta-analyse suggèrent un effet-dose et privilégient aujourd’hui une posologie à 1 mg/kg/14 j. À ce jour, il n’y a pas d’efficacité démontrée des enzymothérapies substitutives sur la prévention des accidents vasculaires cérébraux (pas de passage de la barrière hémato-encéphalique).
Molécule chaperon.6 Depuis 2016, le migalastat, molécule chaperon, est disponible (Galafold : 123 mg/48 h per os à distance des repas). Certaines mutations du gène GLA entraînent la synthèse de protéines dont la structure tertiaire est instable. Le migalastat est un iminosucre qui va occuper le site catalytique de l’αGalA endogène et permettre de stabiliser sa structure jusqu’au lysosome, son lieu d’action. Seules certaines mutations** peuvent être corrigées par le chaperon. Elles ne concernent qu’environ 30 % des malades. La molécule, passant la barrière hémato-encéphalique, pourrait avoir un effet neuroprotecteur central, non démontré à ce jour.
Indication/choix du traitement. Le prix des trois molécules ci-dessus avoisine 200 000 euros/an/patient. Il n’existe pas d’étude comparant les trois traitements entre eux. Les indications actuelles de mise sous traitement spécifiques sont les suivantes :
– en cas de mutation associée à un phénotype classique, tout homme est éligible à un traitement (dès 7 ans en cas de symptômes, à partir de 18 ans si non symptomatique, 16 ans pour le chaperon) ;
– en cas de mutation associée à un phénotype classique, un traitement est débuté chez les femmes en cas d’apparition d’une atteinte d’organe ;
– en cas de mutation associée à un phénotype cardiaque ou non classique, hommes et femmes sont éligibles dès l’apparition d’une atteinte d’organe.
Le délai d’introduction du traitement est essentiel : idéalement précoce, avant le développement de lésions de fibrose irréversible. Le choix d’initier un traitement et la molécule utilisée sont discutés avec le patient. Un autoquestionnaire évaluant les besoins et attentes des patients peut être utile.7 Le traitement est maintenu jusqu’à ce que le rapport bénéfice-risque ne paraisse plus favorable (par exemple : survenue d’une démence vasculaire, d’un cancer, intolérance…).
Molécule chaperon.6 Depuis 2016, le migalastat, molécule chaperon, est disponible (Galafold : 123 mg/48 h per os à distance des repas). Certaines mutations du gène GLA entraînent la synthèse de protéines dont la structure tertiaire est instable. Le migalastat est un iminosucre qui va occuper le site catalytique de l’αGalA endogène et permettre de stabiliser sa structure jusqu’au lysosome, son lieu d’action. Seules certaines mutations** peuvent être corrigées par le chaperon. Elles ne concernent qu’environ 30 % des malades. La molécule, passant la barrière hémato-encéphalique, pourrait avoir un effet neuroprotecteur central, non démontré à ce jour.
Indication/choix du traitement. Le prix des trois molécules ci-dessus avoisine 200 000 euros/an/patient. Il n’existe pas d’étude comparant les trois traitements entre eux. Les indications actuelles de mise sous traitement spécifiques sont les suivantes :
– en cas de mutation associée à un phénotype classique, tout homme est éligible à un traitement (dès 7 ans en cas de symptômes, à partir de 18 ans si non symptomatique, 16 ans pour le chaperon) ;
– en cas de mutation associée à un phénotype classique, un traitement est débuté chez les femmes en cas d’apparition d’une atteinte d’organe ;
– en cas de mutation associée à un phénotype cardiaque ou non classique, hommes et femmes sont éligibles dès l’apparition d’une atteinte d’organe.
Le délai d’introduction du traitement est essentiel : idéalement précoce, avant le développement de lésions de fibrose irréversible. Le choix d’initier un traitement et la molécule utilisée sont discutés avec le patient. Un autoquestionnaire évaluant les besoins et attentes des patients peut être utile.7 Le traitement est maintenu jusqu’à ce que le rapport bénéfice-risque ne paraisse plus favorable (par exemple : survenue d’une démence vasculaire, d’un cancer, intolérance…).
* Association pour les patients de la maladie de Fabry http://www.apmf-fabry.org Association Vaincre les maladies lysosomales https://www.vml- asso.org/-fabry Association pour l’information et la recherche sur les maladies rénales génétiques https://www.airg-france.fr/** Liste est accessible sur www.galafolda menabilitytable.com
Remerciements au Dr Frédéric Barbey pour l’image d’anatomopathologie rénale (fig. 2 ).
Références
1. Lidove O, Kaminsky P, Hachulla E, et al. Fabry disease « the new great imposter »: results of the French Observatoire in Internal Medicine Departments (FIMeD). Clin Genet 2012;81:571‑7.
2. Baig S, Edward NC, Kotecha D, et al. Ventricular arrhythmia and sudden cardiac death in Fabry disease: a systematic review of risk factors in clinical practice. Europace 2018;20:f153-f161.
3. Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab 2018;123:416‑27.
4. Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha-galactosidase. A replacement therapy in Fabry’s disease. N Engl J Med 2001;345:9‑16.
5. Schiffmann R, Kopp JB, Austin HA, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA 2001;285:2743‑9.
6. Hughes DA, Nicholls K, Sunder-Plassmann G, et al. Safety of switching to Migalastat from enzyme replacement therapy in Fabry disease: Experience from the Phase 3 ATTRACT study. Am J Med Genet A 2019;179:1069‑73.
7. Noël E, Dussol B, Lacombe D, Bedreddine N, et al. Treatment needs and expectations for Fabry disease in France: development of a new Patient Needs Questionnaire. Orphanet J Rare Dis 2019;14:284.
2. Baig S, Edward NC, Kotecha D, et al. Ventricular arrhythmia and sudden cardiac death in Fabry disease: a systematic review of risk factors in clinical practice. Europace 2018;20:f153-f161.
3. Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab 2018;123:416‑27.
4. Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha-galactosidase. A replacement therapy in Fabry’s disease. N Engl J Med 2001;345:9‑16.
5. Schiffmann R, Kopp JB, Austin HA, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA 2001;285:2743‑9.
6. Hughes DA, Nicholls K, Sunder-Plassmann G, et al. Safety of switching to Migalastat from enzyme replacement therapy in Fabry disease: Experience from the Phase 3 ATTRACT study. Am J Med Genet A 2019;179:1069‑73.
7. Noël E, Dussol B, Lacombe D, Bedreddine N, et al. Treatment needs and expectations for Fabry disease in France: development of a new Patient Needs Questionnaire. Orphanet J Rare Dis 2019;14:284.
Dans cet article


Résumé
La maladie de Fabry est une maladie rare, monogénique, liée à l’X, où un déficit enzymatique en α-galactosidase A lysosomale provoque l’accumulation intracellulaire de son substrat à l’origine de dysfonctions d’organe. On doit l’évoquer aussi bien chez l’homme que chez la femme, en particulier devant des acroparesthésies à électropyogramme normal, des angiokératomes, une insuffisance rénale familiale ou une cardiopathie hypertrophique possiblement isolée et sans obstacle à l’éjection. Le diagnostic se fait par dosage enzymatique chez l’homme et par analyse génétique chez la femme. Trois traitements sont disponibles : deux enzymothérapies substitutives depuis 2001, une molécule chaperon depuis 2016.