Les maladies à prion, ou encéphalopathies subaiguës spongiformes transmissibles (ESST), sont des affections neurodégénératives touchant aussi bien l’homme que l’animal. Différentes formes sont reconnues chez l’homme : la maladie de Creutzfeldt-Jakob (MCJ) sporadique, les formes génétiques et les formes acquises.
Pathologies dues à un prion, forme anormale d’une protéine
L’agent transmissible est une protéine infectieuse, ou « prion », qui est la forme anormale d’une protéine normale, la PrPc, ancrée sur la membrane des neurones et sensible aux protéases. C’est en acquérant une conformation spatiale anormale que la PrPc est convertie en protéine prion scrapie (PrPsc) et s’accumule dans le cerveau des patients atteints d’ESST. La PrPc est codée, chez l’homme, par le gène PRNP, porté par le chromosome 20. Un polymorphisme du codon 129 s’exprime chez tout individu, définissant trois génotypes possibles : méthionine/méthionine (M/M) pour environ 40 % de la population générale, valine/valine (V/V) pour 10 % ou méthionine/valine (M/V) pour 50 %. La sensibilité aux ESST ainsi que l’expression clinique ou neuropathologique de la maladie varient en fonction de ce génotype.
Maladies très rares et sporadiques le plus souvent
Ce sont des maladies rares dont l’incidence, toutes formes confondues, se situe autour de deux cas par million. La forme sporadique représente 90 % des ESST.
La recherche des facteurs de risque de la MCJ sporadique a fait l’objet de plusieurs études cas-témoins qui ont exploré les habitudes alimentaires, les professions, les contacts avec les animaux, les antécédents médicaux.1,2 Ces études n’ont pas révélé de facteur de risque. Une étude récente est en faveur d’un effet « cohorte », suggérant qu’une exposition à un facteur environnemental pourrait jouer un rôle dans l’apparition de la MCJ sporadique.3
Environ 10 % des maladies à prion sont associées à des mutations du gène PRNP. Il existe trois phénotypes : les MCJ génétiques, le syndrome de Gerstmann-Sträussler-Scheinker (SGSS) et l’insomnie fatale familiale (IFF).
Les MCJ acquises regroupent le kuru (disparu grâce à l’interdiction du cannibalisme), les MCJ iatrogènes par contamination cérébrale ou juxtacérébrale (greffes de cornée, interventions neurochirurgicales et surtout greffes de dure-mère) ou périphérique (traitement par hormone de croissance d’origine humaine) et la variante de la MCJ (vMCJ).
Les cas de MCJ par greffes de dure-mère ont été observés surtout au Japon et ceux liés à l’hormone de croissance surtout en France. Le nombre de cas de MCJ iatrogène a diminué depuis la généralisation de l’utilisation de la dure-mère synthétique en 1994 et de l’hormone de croissance recombinante en 1988.
La vMCJ a été décrite en 19964 et son lien avec l’encéphalopathie spongiforme bovine (ESB) prouvé sur des arguments épidémiologiques et expérimentaux. D’un point de vue épidémiologique, la vMCJ se distingue par deux caractéristiques : la distribution géographique, corrélée à l’exposition à l’agent de l’ESB et un jeune âge au début de la maladie. Les recherches sur la voie de transmission de l’agent de l’ESB à l’homme ont privilégié l’hypothèse alimentaire. En 2024, on dénombre 178 cas au Royaume-Uni dont trois cas après transfusion de culots globulaires non déleucocytés et 29 cas en France dont deux cas professionnels décédés en 20195 et 2021. Les derniers patients atteints de vMCJ d’origine alimentaire sont décédés en 2016 au Royaume-Uni et en 2014 en France.
Diagnostic des différentes formes cliniques
MCJ sporadique
L’âge moyen de survenue de cette forme est de 69 ans ; le sex-ratio est égal à 1 et l’évolution est rapidement fatale en sept mois.6
Signes cliniques
Une phase de prodromes (asthénie, anxiété, insomnie…) est classique. La phase d’état peut débuter de manière brutale (pseudovasculaire) ou plus souvent en quelques jours. La maladie affecte le cerveau de façon diffuse, expliquant les signes neurologiques variés. La démence reste le signe cardinal associant troubles de mémoire, de l’orientation temporelle ou spatiale, du langage, des gestes ou de la reconnaissance. Les myoclonies spontanées ou provoquées sont souvent diffuses. Le syndrome cérébelleux se manifeste surtout par l’ataxie mais peut toucher les membres supérieurs ou entraîner nystagmus et dysarthrie. Les troubles visuels vont de la simple gêne aux hallucinations ou à la cécité corticale. Le syndrome pyramidal peut se résumer à des réflexes trop vifs. Les signes extrapyramidaux sont variés : hypertonie, dystonie, tremblement, mouvements anormaux. L’évolution se fait dans 64 % des cas vers un mutisme akinétique.6
Examens complémentaires
Les résultats usuels sanguins et du liquide céphalo-rachidien (LCR) sont normaux.
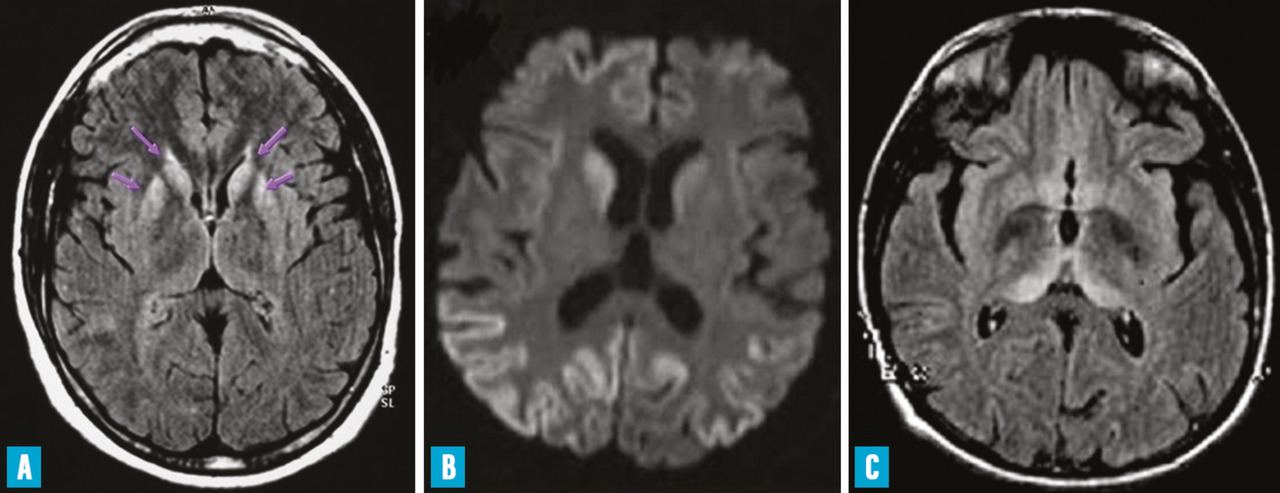
L’observation sur l’imagerie par résonance magnétique (IRM) d’hypersignaux dans les noyaux gris centraux ou dans le cortex cérébral, sur les séquences T2, FLAIR ou de diffusion avec une diminution du coefficient de diffusion apparent de l’eau (ADC) est fréquente (fig. 1 A et B). Il n’existe pas de prise de contraste.
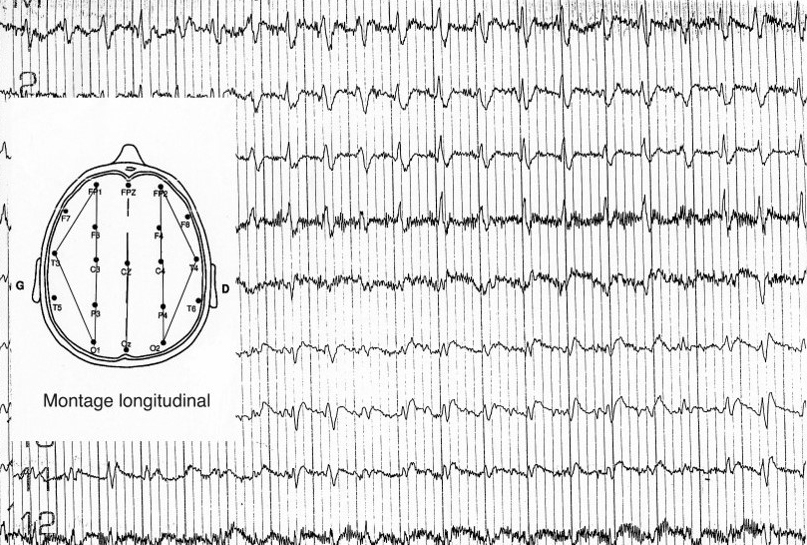
L’électroencéphalogramme (EEG) de veille est en règle anormal : ralentissement du rythme de base, décharges d’ondes lentes polymorphes répétitives et, dans 60 % des cas, pointes-ondes lentes bi- ou triphasiques paroxystiques, diffuses et périodiques définissant l’EEG caractéristique (fig. 2).
La détection de la protéine 14 - 3 - 3 dans le LCR est un test sensible et spécifique.7 Elle reflète, comme l’augmentation importante et isolée de la protéine Tau totale, la dégénérescence neuronale. La recherche d’une faible quantité de PrPsc dans le LCR est possible, par une technique d’amplification du « protein misfolding » appelée real-time quaking-induced conversion (RT-QuIC)8 qui a une grande valeur diagnostique (sensibilité 90 % et spécificité 99 %).
L’étude de PRNP élimine une anomalie génétique et détermine le polymorphisme du codon 129. L’homozygotie M/M est plus fréquente dans la MCJ sporadique (70 %) que dans la population générale (40 %) et constitue un facteur de risque.
Le diagnostic de certitude ne peut être obtenu qu’avec l’étude du tissu cérébral après autopsie. Celle-ci permet d’observer les trois lésions caractéristiques : gliose, spongiose et raréfaction neuronale. Par les techniques d’immunocytochimie, des dépôts ou des amas de PrPsc sont visualisés. Les plaques amyloïdes sont rares. L’étude de la migration de la PrPsc par western blot se fait à partir de fragments cérébraux congelés.
Les variations du phénotype clinique en fonction du polymorphisme du codon 129 et du profil de migration de la PrPsc (type 1 ou 2A) en western blot permettent de définir différentes formes cliniques probablement sous-tendues par des souches différentes de prion.9
Formes génétiques à transmission autosomique dominante
Elles sont liées à différentes mutations ou à des insertions de nucléotides entre les codons 51 et 91 du gène PRNP. La transmission est toujours autosomique dominante. L’histoire familiale peut manquer, soulignant l’importance d’effectuer l’étude du gène PRNP.
Pour l’ensemble des ESST génétiques, la fréquence des EEG caractéristiques, de la détection de la protéine 14 - 3 - 3 ou de la positivité de la RT-QuIC est variable en fonction de la forme. Un tracé EEG périodique et une protéine 14 - 3 - 3 positive sont fréquents en cas de mutation 200, rares en cas de SGSS et d’IFF.
Maladies de Creutzfeldt-Jakob génétiques
Les MCJ génétiques sont dues à des mutations du gène PRNP pouvant être situées sur différents codons : 200, 178, 180, 210... Elles peuvent aussi être dues à des insertions d’un petit nombre de nucléotides. Le tableau clinique dépend de la mutation. Les signes cliniques de la forme avec mutation 200 – qui est la plus fréquente (65 % des cas) – sont comparables à ceux décrits pour la forme sporadique. Les autres mutations ont en général été découvertes de manière fortuite chez des patients présentant un tableau de MCJ.
Syndrome de Gerstmann-Sträussler-Scheinker
Toujours d’origine génétique, sa dénomination particulière est liée aux particularités cliniques, évolutives (sur plusieurs années) et neuropathologiques (présence de plaques de PrP particulières : multicentriques). La mutation 102, la plus fréquente, est responsable de la forme ataxique. La mutation 117, plus rare, se traduit par un syndrome pyramidal ou pseudobulbaire accompagné d’une démence plus ou moins précoce. D’autres mutations ou des insertions d’un nombre élevé de nucléotides peuvent causer un SGSS.
Insomnie fatale familiale
L’IFF est liée à une mutation ponctuelle du codon 178 associée, sur le même allèle, à un codon 129 codant une méthionine (si le codon 129 code une valine, le tableau clinique est celui d’une MCJ génétique). Cliniquement, elle est caractérisée par l’association d’une insomnie sévère (avec hallucinations), de troubles végétatifs (disparition des rythmes circadiens, hyperactivité sympathique, troubles sphinctériens), de difficultés motrices. La démence peut être tardive. Les myoclonies sont rares. L’électroencéphalogramme de sommeil est anormal. La durée de la maladie varie entre quatre et vingt-quatre mois. Le phénotype neuropathologique est particulier : gliose et peu de spongiose localisées surtout dans les noyaux antérieurs et dorso-médians du thalamus.
Trois types de formes acquises
Maladie de Creutzfeldt-Jakob iatrogène par contamination de proximité cérébrale
La durée d’incubation est en moyenne de six ans. Le tableau clinique est proche de celui de la MCJ sporadique. L’EEG et la détection de la protéine 14 - 3 - 3 sont souvent positifs.
MCJ iatrogène liée à un traitement par hormone de croissance
La maladie débute généralement par une ataxie cérébelleuse, des troubles oculomoteurs et un nystagmus. Il existe souvent un tremblement, une prise de poids, des troubles du sommeil. Ce n’est qu’après quelques mois d’évolution qu’apparaissent les signes pyramidaux, les myoclonies et la démence. La durée moyenne d’évolution est de dix-huit mois. Le tableau clinique est plutôt stéréotypé. Les anomalies EEG sont discrètes. La détection de la protéine 14 - 3 - 3 est inconstamment positive.
Variante de la MCJ
La vMCJ débute par des troubles psychiatriques (dépression sévère, état délirant, hallucinations…) ou des douleurs. Le diagnostic n’est évoqué que quelques semaines plus tard, quand apparaissent l’ataxie cérébelleuse, le syndrome pyramidal ou les myoclonies. L’atteinte intellectuelle est plus tardive. L’évolution se fait vers le mutisme akinétique. Le décès survient après quinze mois en moyenne. L’EEG n’est pas périodique. La protéine 14 - 3 - 3 n’est détectable que dans la moitié des cas ; la RT-QuIC est en règle négative. Des hypersignaux sur l’IRM cérébrale sont fréquemment observés dans les noyaux pulvinar et dorsomédian du thalamus ; ils ont une importante valeur diagnostique (fig. 1C).10 En raison de son accumulation dans les organes lymphoïdes, la PrPsc peut être mise en évidence sur une biopsie d’amygdale pharyngée. L’étude du gène PRNP ne décèle pas de mutation, tous les patients sont M/M au codon 129 excepté le dernier patient britannique décédé en 2016 qui était M/V. L’étude neuropathologique permet d’observer des plaques PrPsc particulières dites florides (plaques amyloïdes entourées de spongiose). Le profil de migration en western blot de la PrPsc est identique à celui de la PrPsc bovine (type 2B).
Prise en charge globale sans traitement curatif
Plan national d’accompagnement
La circulaire DGS/DHOS/DGAS/DSS n° 2001 - 139 du 14 mars 2001 entérine la création de la cellule nationale de référence des MCJ qui a pour mission d’aider au diagnostic et à la prise en charge des patients et de leur famille et qui joue aussi un rôle d’information générale sur les ESST. Ce n’est pas un lieu de prise en charge directe des patients ; celle-ci doit se faire à proximité de leur domicile.
Traitements médicaux de confort
Les traitements dits « de confort » sont les suivants : antalgiques en cas de douleur, benzodiazépines ou antiépileptiques contre les myoclonies, hydratation, prévention et traitement des complications de décubitus.
Aucun traitement n’a fait aujourd’hui la preuve de son efficacité. Avant les années 2000, de nombreux médicaments ont été testés en dehors de tout protocole. Depuis, des protocoles thérapeutiques ont été mis en place.
Ainsi, la quinacrine dont l’efficacité n’avait pas été montrée en utilisation compassionnelle en France11 a fait l’objet d’essais thérapeutiques en ouvert au Royaume-Uni et en double aveugle aux États-Unis sans effet positif sur la survie.12,13
Le polysulfate de pentosane par voie intraventriculaire a été à l’étude dans plusieurs pays (Japon, Royaume-Uni et France). Un effet est possible dans la vMCJ puisque sur cinq patients traités au Royaume-Uni, quatre ont eu une survie plus longue que celle des patients non traités.14
En raison de résultats expérimentaux et d’essais compassionnels ouverts encourageants, une étude randomisée en double aveugle versus placebo a été mise en place en Italie et en France pour tester l’efficacité de 100 mg/j de doxycycline. Cette étude, dont le résultat sur la survie a été négatif, a montré qu’une étude randomisée en double aveugle contre placebo était possible dans une maladie rapidement mortelle et a souligné la limite des essais ouverts.15
Des données expérimentales suggèrent que l’utilisation d’antisens pour limiter l’expression de la PrPc permettrait de réduire la réplication de l’agent et de bloquer partiellement les processus de neurodégénérescence PrPc-dépendants induits par les prions. Un essai thérapeutique de phase I-IIa multicentrique international a débuté en 2024.
Maladies à prion rares et d’expression variée
Les données épidémiologiques et cliniques des maladies à prion sont variables d’une forme à l’autre. Elles constituent cependant un groupe de maladies bien individualisées par leur rareté, leur caractère transmissible et les anomalies neuropathologiques et biochimiques qui leur sont liées.
2. Collins S, Law MG, Fletcher A, et al. Surgical treatment and risk of sporadic Creutzfeldt-Jakob disease: A case-control study. Lancet 1999;353(9154):693-7.
3. Denouel A, Brandel JP, Seilhean D, et al. The role of environmental factors on sporadic Creutzfeldt-Jakob disease mortality: Evidence from an age-period-cohort analysis. Eur J Epidemiol 2023;38(7):757-64.
4. Will RG, Ironside JW, Zeidler M, et al. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 1996;347(9006):921-5.
5. Brandel JP, Vlaicu MB, Culeux A, et al. Variant Creutzfeldt-Jakob disease diagnosed 7.5 years after occupational exposure. N Engl J Med 2020;383(1):83-5.
6. Denouel A, Brandel JP, Peckeu-Abboud L, et al. Prospective 25-year surveillance of prion diseases in France, 1992 to 2016: A slow waning of epidemics and an increase in observed sporadic forms. Euro Surveill 2023;28(50):2300101.
7. Zerr I, Bodemer M, Geffeler O, et al. Detection of 14-3-3 protein in the cerebrospinal fluid supports the diagnosis of Creutzfeldt-Jakob disease. Ann Neurol 1998;43(1):32-40.
8. Atarashi R, Satoh K, Sano K, et al. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat Med 2011;17(2):175-8.
9. Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999;46(2):224-33.
10. Zeidler M, Sellar RJ, Collie DA, et al. The pulvinar sign on magnetic resonance imaging in variant Creutzfeldt-Jakob disease. Lancet 2000;355(9213):1412-8.
11. Haik S, Brandel JP, Salomon D, et al. Compassionate use of quinacrine in Creutzfeldt-Jakob disease fails to show significant effects. Neurology 2004;63(12):2413-5.
12. Collinge J, Gorham M, Hudson F, et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): A patient-preference trial. Lancet Neurol 2009;8(4):334-44.
13. Geschwind MD, Kuo AL, Wong KS, et al. Quinacrine treatment trial for sporadic Creutzfeldt-Jakob disease. Neurology 2013;81(23):2015-23.
14. Newman PK, Todd NV, Scoones D, Mead S, et al. Postmortem findings in a case of variant Creutzfeldt-Jakob disease treated with intraventricular pentosan polysulfate. J Neurol Neurosurg Psychiatry 2014;85(8):921-4.
15. Haik S, Marcon G, Mallet A, et al. Doxycycline in Creutzfeldt-Jakob disease: A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol 2014;13(2):150-8.