Connaître la démarche diagnostique en présence d’une gammapathie monoclonale.
Définition et épidémiologie
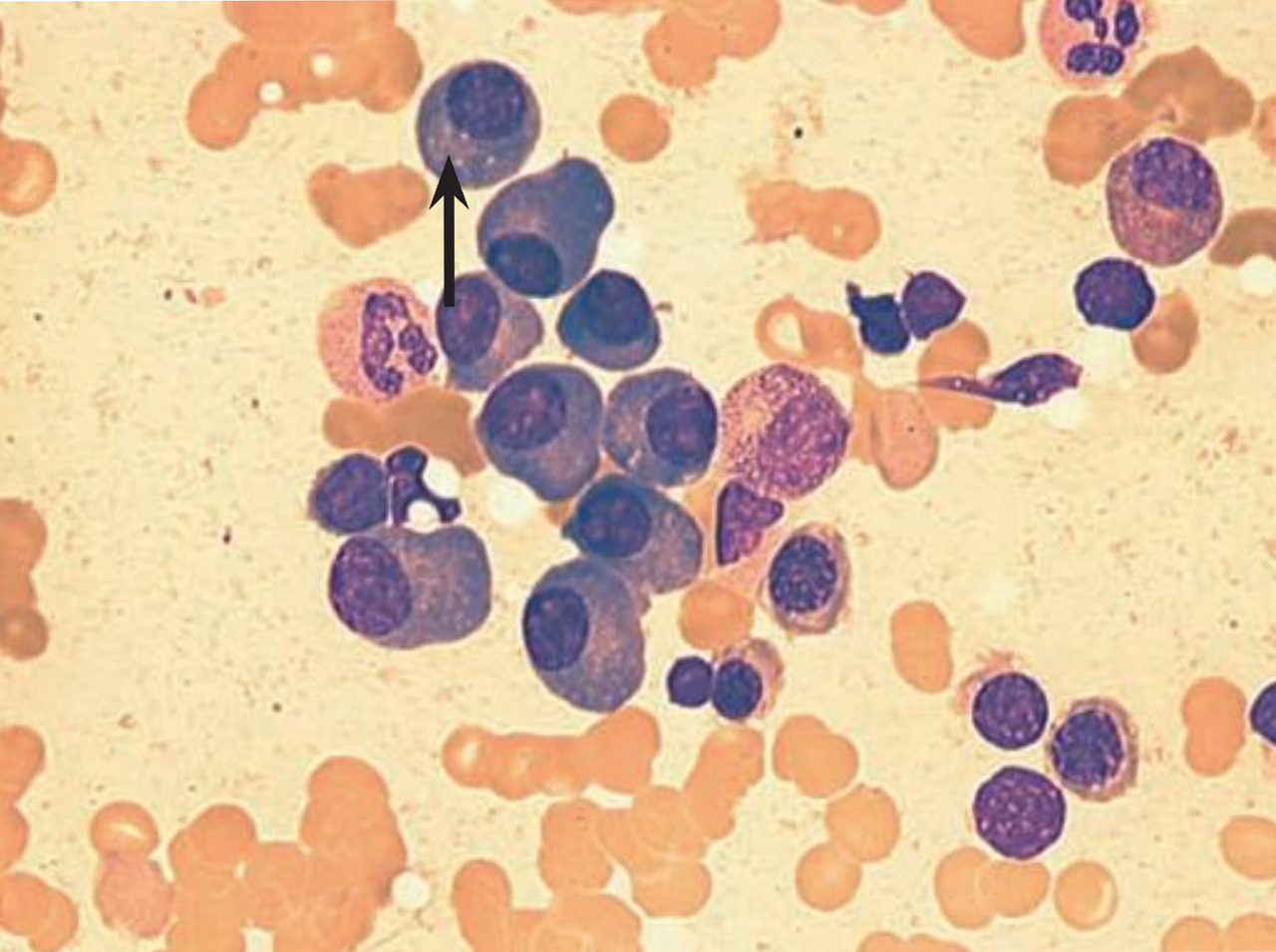
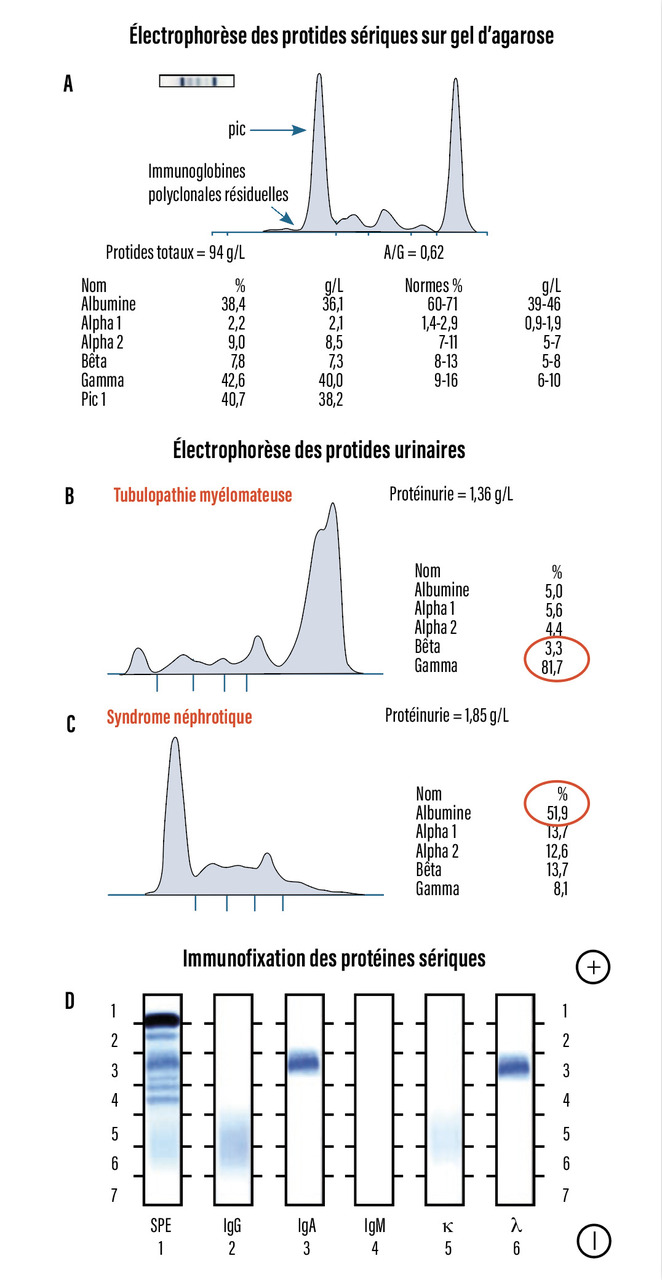
Le myélome multiple des os est une hémopathie lymphoïde B mature qui associe, par définition, une prolifération plasmocytaire tumorale médullaire et, le plus souvent, la présence d’une immunoglobuline monoclonale, complète ou incomplète, issue de ces plasmocytes clonaux. Le diagnostic repose sur la mise en évidence de ces deux composantes : la prolifération plasmocytaire, par l’examen du frottis médullaire après aspiration (fig. 1), et la gammapathie monoclonale, par les examens immuno-hématologiques sanguins et urinaires (fig. 2).
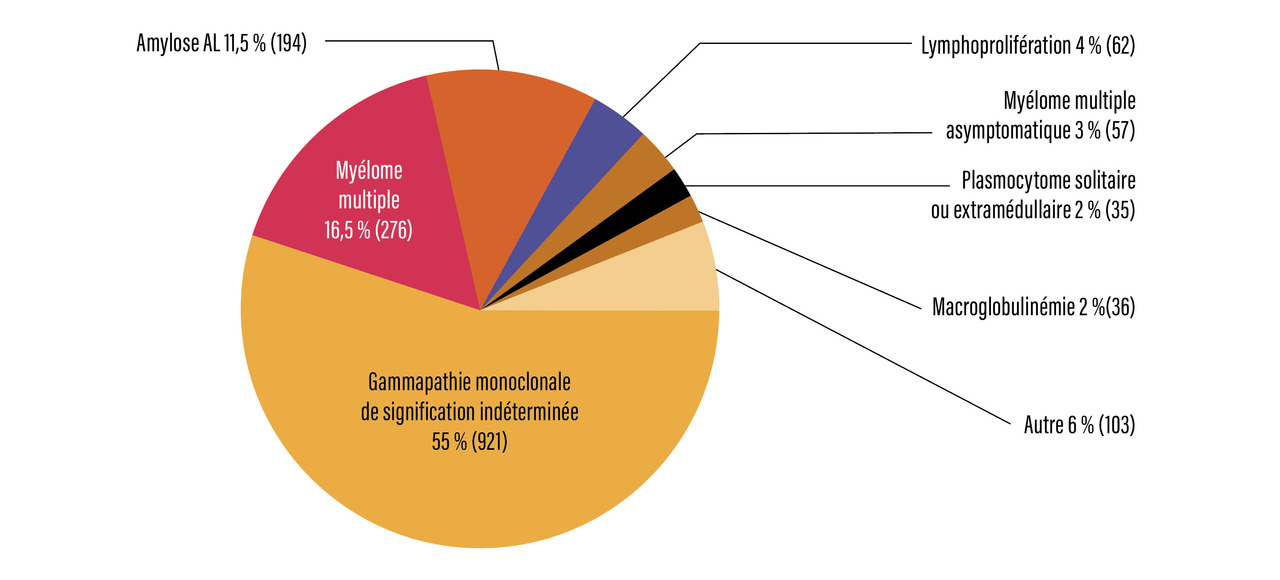
Le myélome représente 1 % des cancers et 12 % des hémopathies malignes. Son incidence est estimée à 4,2 chez l’homme et à 2,9 chez la femme pour 100 000 habitants par an, avec 5 442 nouveaux cas en 2018 (Santé publique France). La médiane d’âge au diagnostic est de 70 ans chez l’homme, 74 chez la femme, avec un sex-ratio de 1,4. Les deux tiers des patients ont 65 ans ou plus. La cause est inconnue, mais le myélome est associé à une exposition aux radiations ionisantes et l’utilisation de pesticides a été incriminée. Il est quasi constamment précédé d’une gammapathie monoclonale de signification indéterminée (monoclonal gammopathy of undetermined significance [MGUS] des Anglo-Saxons). L’incidence de cette dernière augmente avec l’âge, pour atteindre 10 % au-dessus de 80 ans. Le taux d’évolution des gammapathies monoclonales de signification indéterminée vers une maladie lymphoproliférative est de 1 % par an.
Diagnostic
Circonstances de découverte
Le myélome peut être découvert de façon fortuite lors de la réalisation d’examens de routine : accélération de la vitesse de sédimentation contrastant avec une CRP normale (absence de syndrome inflammatoire), hyperprotidémie, présence de rouleaux sur le frottis sanguin. Le diagnostic peut être évoqué devant des symptômes cliniques en rapport avec la prolifération tumorale (douleurs osseuses, hypercalcémie, anémie, compression neurologique), la présence de l’immunoglobuline monoclonale (insuffisance rénale) ou la diminution des immunoglobulines polyclonales (immunoparésie) responsable d’un déficit immunitaire humoral avec infections à germes encapsulés (pneumopathies à pneumocoque).
Examens immuno-hématologiques
L’immunoglobuline monoclonale est suspectée sur la présence d’une bande étroite migrant dans les gammaglobulines ou, moins souvent, dans la région des bêtaglobulines, sur l’électrophorèse des protides sériques (EPS) ou urinaires (EPU) (fig. 2A). L’électrophorèse des protides sériques ou urinaires permet, par intégration du pic (aire sous la courbe), d’estimer la quantité d’immunoglobuline monoclonale. Seules l’immunofixation (IF) ou l’immuno-électrophorèse (IEP) permettent d’identifier l’immunoglobuline monoclonale en déterminant sa chaîne lourde (IgG dans 60 % des cas ou IgA dans 20 % des cas) et sa chaîne légère (kappa ou lambda) [fig. 2B]. Parfois, l’électrophorèse des protides sériques ne montre pas de pic mais une hypogammaglobulinémie (15 à 20 % des myélomes) : il faut alors savoir rechercher la présence de chaînes légères isolées kappa (deux tiers des cas) ou lambda (un tiers des cas), dans le sang ou dans les urines, par immunofixation ou immuno-électrophorèse. La présence de chaînes légères dans les urines correspond à ce que l’on nommait protéinurie de Bence-Jones, détectée par des méthodes de précipitation à chaud à l’acide sulfosalicylique. Dans moins de 1 % des cas, aucune immunoglobuline monoclonale n’est détectée : il s’agit d’un myélome non sécrétant ou non excrétant. Le diagnostic est évoqué devant une hypogammaglobulinémie à l’électrophorèse des protides sériques et confirmé par la plasmocytose médullaire associée à un critère CRAB (v. infra).
Critères diagnostiques
Le diagnostic de myélome repose sur la présence d’une infiltration plasmocytaire tumorale au myélogramme au moins égale à 10 % associée ou non à une immunoglobuline monoclonale. S’il existe des symptômes cliniques (anémie, signes osseux, insuffisance rénale ou hypercalcémie), le myélome est dit symptomatique (tableau 1). La présence d’un critère CRAB (C = hyperCalcémie, R = insuffisance Rénale, A = Anémie, B = Bone = os) constitue un argument de traitement pour l’International Myeloma Working Group (IMWG) (tableau 2a).
Bilan pronostique
Le diagnostic de myélome multiple nécessite la réalisation d’examens qui permettent d’en établir le stade et d’évaluer son pronostic avant la prise en charge thérapeutique.
Sur le plan biologique : hémogramme, clairance de la créatininémie, calcémie, protidémie, albuminémie, uricémie, bilan hépatique, protéine C réactive (CRP), bêta- 2 -microglobulinémie, myélogramme avec analyse cytogénétique. L’étude des plasmocytes tumoraux purifiés par hybridation in situ permet de détecter des anomalies de mauvais pronostic telles que la del(17p) ou la t(4 ;14) ou les anomalies du 1 (del1q,+ 1p).
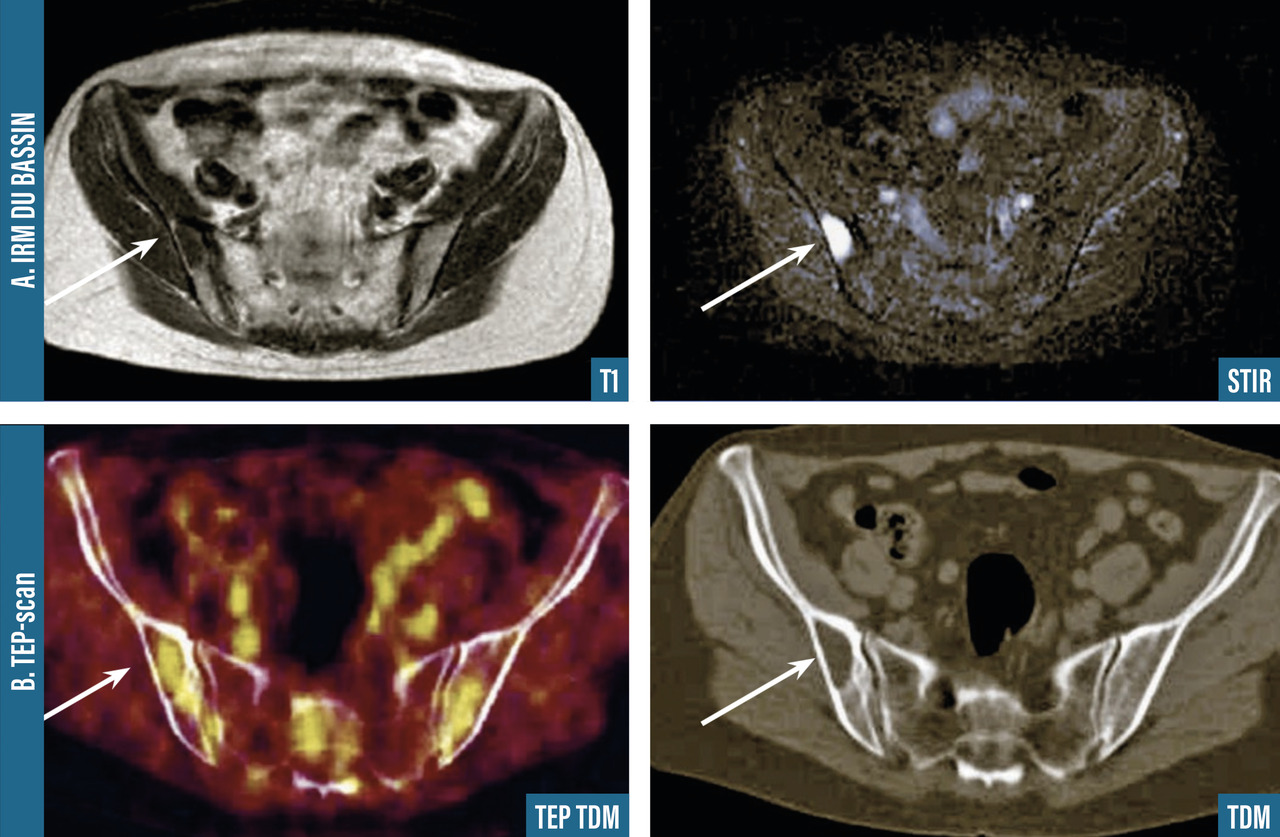
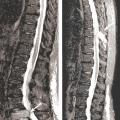
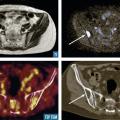
Sur le plan morphologique : l'imagerie osseuse par radiographies standard du squelette (crâne, rachis cervico-dorso-lombaire, bassin, os longs) doit être remplacée par le scanner faible dose, beaucoup plus sensible et dont l'irradiation est plus faible ; l'imagerie par résonance magnétique (IRM) axiale (bassin et rachis) ou, mieux, du corps entier, est utile en cas de radiographies standard normales pour rechercher une infiltration nodulaire méconnue ou une infiltration diffuse ; elle permet, en outre, de dépister une épidurite tumorale avec risque de compression médullaire. Le TEP-scan associe morphologie et imagerie fonctionnelle et permet le suivi chirurgical orthopédique. Les tumeurs plasmocytaires ne fixent pas le Tc99m : la scintigraphie osseuse est donc inutile au cours du myélome.
Au terme de ce bilan, le myélome peut être classé selon la classification de Salmon et Durie (tableau 3a). Actuellement, seuls les patients avec un myélome symptomatique (présence d’un critère CRAB ou SLIM-CRAB [tableau 2b]) sont traités. L’indice pronostique international (International Staging System [ISS]) est un critère pronostique mais non de traitement (tableau 3b). L’ISS révisé ou ISS-R incorpore la LDH et les anomalies cytogénétiques (tableau 3c).
Diagnostics différentiels
Devant toute gammapathie monoclonale non IgM
La présence d’une immunoglobuline monoclonale IgM évoque avant tout une macroglobulinémie de Waldenström, qui correspond à un lymphome lymphoplasmocytaire dont le tableau clinique comporte une anémie et un syndrome tumoral (hépatosplénomégalie et adénopathies).
La présence d’une immunoglobuline monoclonale non IgM peut être en rapport avec une gammapathie monoclonale de signification indéterminée, un myélome indolent ou un myélome symptomatique : la réalisation du myélogramme, de l’imagerie et des examens biologiques simples (hémogramme, créatininémie, calcémie) permet de faire la différence.
Dans les cas difficiles, une gammapathie monoclonale de signification indéterminée peut être associée à des tassements vertébraux d’origine ostéoporotique chez une femme âgée ménopausée. L’IRM permet de différencier un tassement vertébral ostéoporotique d’une localisation vertébrale tumorale.
Formes cliniques particulières
Plasmocytome solitaire
Le diagnostic repose sur l’absence de plasmocytose médullaire, de lésions osseuses (lésion unique à l’IRM corps entier ou au TEP-scan) et de critères CRAB. La localisation la plus fréquente est osseuse, mais il existe des formes extra-osseuses, notamment de la sphère ORL. Le traitement est local et repose sur la radiothérapie. Le suivi doit être prolongé, l’évolution se faisant dans la moitié des cas vers un myélome multiple en l’espace de cinq ans.
Leucémie à plasmocytes
Elle se traduit par une plasmocytose sanguine d’au moins 2G/L ou 20 % (toutefois la dernière définition de l’IMWG a abaissé le seuil à 5 % de plasmocytes circulants) et une présentation clinique de leucémie aiguë avec cytopénies et altération de l’état général.
Son pronostic reste sombre malgré les traitements actuels et le recours aux traitements intensifs. Elle peut être inaugurale ou survenir en cours de l’évolution terminale du myélome multiple (leucémie à plasmocytes secondaire).
Amylose AL
En rapport avec le dépôt tissulaire organisé de chaînes légères (plus souvent lambda que kappa) alors que la prolifération plasmocytaire est le plus souvent négligeable. La symptomatologie est variée et il faut savoir l’évoquer devant une neuropathie périphérique, un canal carpien, un purpura périorbitaire, un syndrome néphrotique, une macroglossie. C’est l’atteinte cardiaque, sous la forme d’une cardiopathie hypertrophique à fonction systolique longtemps conservée, qui en grève le pronostic. Le diagnostic repose sur l’histologie d’un organe atteint : mise en évidence d’un dépôt périvasculaire amorphe coloré par le rouge Congo avec biréfringence jaune-vert en lumière polarisée et identification de la chaîne légère lambda ou kappa en immuno-histochimie. Le traitement des formes avec atteinte cardiaque est une urgence.
POEMS
Le diagnostic requiert la présence d’une polyneuropathie et d’une gammapathie monoclonale, les autres symptômes (organomégalie, endocrinopathie et atteinte cutanée = Skin) étant inconstants. Il est confirmé par l’élévation du taux de VEGF sanguin. Les lésions osseuses sont le plus souvent ostéocondensantes.
Autres
Une gammapathie monoclonale peut aussi être satellite d’un lymphome, d’une myélodysplasie (fig. 3), et surtout d’une atteinte rénale, neurologique ou cutanée (gammapathie de signification clinique [MGCS])
Manifestations cliniques et complications
Atteinte osseuse
Clinique et physiopathologie
Elle est présente dans 70 % des cas au diagnostic mais peut aussi survenir en cours d’évolution. Les douleurs osseuses intéressent habituellement le squelette axial (rachis, côtes, bassin). D’aggravation progressive, elles nécessitent le recours aux antalgiques majeurs et ont un retentissement fonctionnel pouvant confiner le patient au lit. Les tuméfactions des os plats (crâne, sternum) et les fractures pathologiques révélatrices sont moins fréquentes. La maladie osseuse du myélome est en rapport avec un découplage du remodelage osseux : le recrutement et l’hyperactivité des cellules ostéoclastiques, sous l’influence de l’IL- 6, produite par le micro-environnement médullaire et les plasmocytes tumoraux eux-mêmes, entraînent une augmentation de la destruction osseuse par hyperexpression du RANK-L se liant au RANK (récepteur activateur de la voie Nf-kB appartenant à la superfamille des TNF), alors que l’ostéoprotégérine (OPG), qui bloque le RANK, est diminuée avec augmentation du rapport RANK-L/OPG. Les cellules ostéoblastiques sont au contraire déprimées, par blocage de la voie Wnt/DKK1 responsable de la maturation des ostéoblastes, avec une balance négative de l’ostéoformation par rapport à l’ostéolyse.
Imagerie du myélome
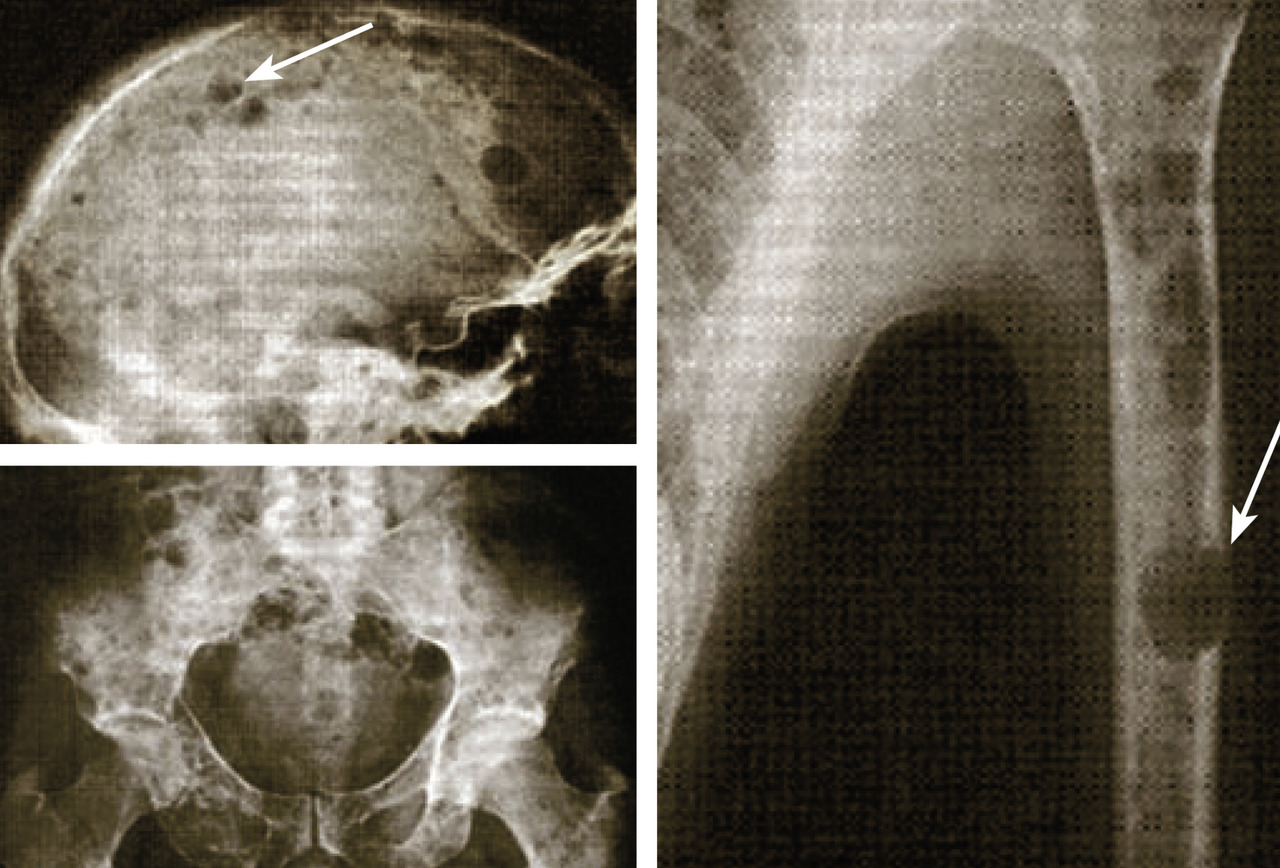
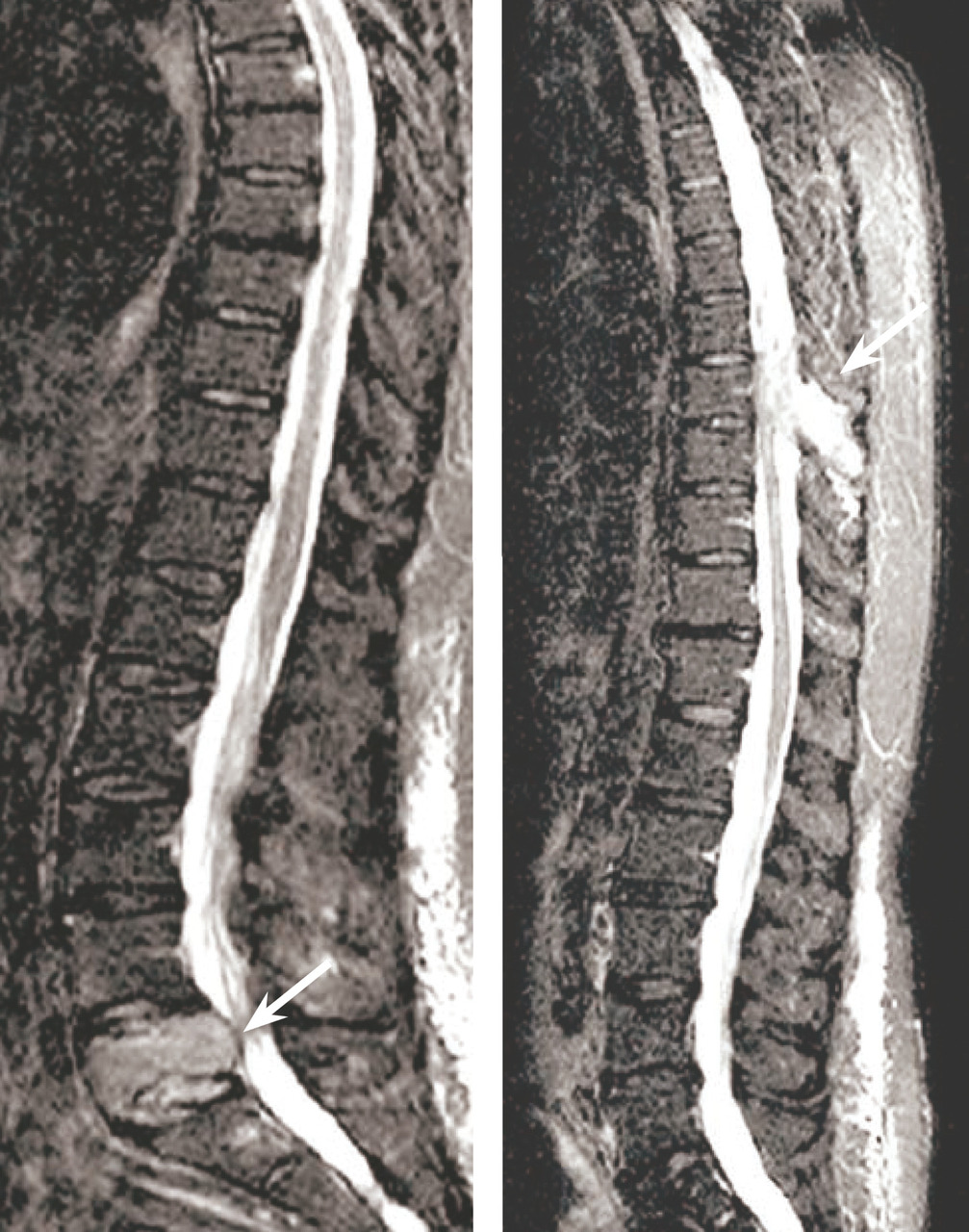
Les clichés standard du squelette axial (crâne, rachis, côtes et bassin) restent encore la référence pour diagnostiquer une atteinte osseuse mais devraient être remplacés, en raison de leur faible sensibilité, par l’imagerie moderne. Les anomalies évocatrices sont une lyse à l’emporte-pièce (géode ou lacune), visible sur les os plats, associée ou non à une déminéralisation diffuse, ou sur les os longs avec résorption corticale et menace de fracture pathologique (fig. 4). Les lacunes peuvent être totalement asymptomatiques, notamment lorsqu’elles siègent sur le crâne. Les tassements vertébraux, plus ou moins complets, voire en galette, sont toujours symptomatiques. Le scanner du corps entier faible dose devrait remplacer les clichés standard en raison de sa plus grande sensibilité, de sa durée de réalisation plus courte et de sa plus faible dose délivrée d’irradiation. L’IRM du corps entier et le TEP-scan détectent 20 % de lésions supplémentaires par rapport aux clichés osseux standard sous la forme d’images nodulaires en hyposignal T1 et hypersignal T2 et STIR, pour l’IRM (fig. 5), ou de foyers hypermétaboliques correspondant aux lacunes sur le scanner, pour le TEP-scan (fig. 6). Ces examens sont particulièrement utiles dans les formes peu avancées de la maladie (absence d’anomalie sur les clichés standard), pour dépister une épidurite ou confirmer le caractère tumoral du tassement vertébral (IRM) et pour suivre l’évolution sous traitement (réponse métabolique au TEP-scan avec disparition des foyers hypermétaboliques). Leur utilisation est en cours de validation et limitée par leur accessibilité.
Atteinte rénale
Elle est très fréquente : la protéinurie est détectée dans 80 % des cas au diagnostic, en dehors d’un myélome à chaînes légères. L’insuffisance rénale aiguë oligo-anurique peut être révélatrice ; elle est présente dans 30 % des cas au diagnostic et dans la moitié des cas au cours de l’évolution. Elle est liée, dans 90 % des cas, à la précipitation des chaînes légères kappa ou lambda dans le tube contourné distal, après liaison à la protéine physiologique de Tamm-Horsfall (uromoduline).
La tubulopathie myélomateuse complique les myélomes avec élimination urinaire de chaînes légères. Les facteurs favorisant la concentration urinaire de chaînes légères libres et leur précipitation sont une déshydratation et une acidification des urines qui peut être déclenchée par une infection, une hypercalcémie, un médicament néphrotoxique (anti-inflammatoires non stéroïdiens, aminosides, produit de contraste iodé). Si une histologie rénale est réalisée, elle montre la précipitation des chaînes légères (kappa ou lambda en immunohistochimie) sous la forme de cylindres dans les tubules avec une réaction inflammatoire.
Plus rarement, l’atteinte rénale peut comporter une amylose caractérisée par un dépôt amorphe, d’organisation fibrillaire, de chaînes légères dans les capillaires glomérulaires et se traduisant par une protéinurie glomérulaire avec plus de 30 % d’albumine à l’électrophorèse des protides urinaires.
La maladie des dépôts d’immunoglobuline monoclonale, ou maladie de Randall, est encore plus rare et est due à un dépôt non organisé de chaînes lourdes ou légères (le plus souvent kappa) au niveau des membranes basales.
Enfin, le syndrome de Fanconi est exceptionnel : il est dû à l’accumulation intracytoplasmique de cristaux constitués de chaînes légères dans les cellules du tube contourné proximal ; il se manifeste par une acidose métabolique avec hypokaliémie, glycosurie normoglycémique, hypo-uricémie avec hyperuraturie, hypophosphatémie avec hyperphosphaturie et amino-acidurie.
Complications neurologiques
Elles sont dominées par la compression médullaire, liée à un tassement vertébral avec recul du mur postérieur, ou à une épidurite associée ou isolée. La symptomatologie initiale peut être fruste, avec un déficit moteur modéré, masqué par les rachialgies. Le tableau complet associe un déficit moteur avec syndrome pyramidal et troubles sphinctériens. Il s’agit d’une urgence thérapeutique. La compression peut être radiculaire avec un tableau de cruralgie ou de sciatique d’horaire inflammatoire. Les neuropathies périphériques sont exceptionnelles, hors POEMS.
Hypercalcémie
Une calcémie supérieure à 2,75 mmol/L est présente dans 30 % des cas au diagnostic et nécessite une hospitalisation en urgence. Elle peut être masquée par une hypo-albuminémie et son taux doit alors être corrigé en fonction de l’albumine. Elle est due à une libération accrue du calcium osseux par l’ostéolyse, conjointement à la diminution de l’excrétion rénale en raison de l’atteinte tubulaire myélomateuse. Elle peut se manifester par une déshydratation, une confusion et des troubles digestifs (nausées, vomissements, douleurs abdominales).
Insuffisance médullaire
L’anémie est très fréquente au cours du myélome et souvent révélatrice. Elle est due à l’envahissement médullaire mais aussi à une action spécifique des plasmocytes tumoraux qui stimulent l’apoptose des érythroblastes médullaires par la voie du ligand Fas. Elle peut être faussement majorée par l’hémodilution liée à l’hyperprotidémie. Elle se manifeste par une asthénie d’aggravation progressive, une pâleur et une dyspnée d’effort. Le frottis sanguin montre le phénomène caractéristique des rouleaux avec empilement des hématies en pile d’assiettes lié à l’agrégation de l’immunoglobuline monoclonale adsorbée à leur surface. La neutropénie et la thrombopénie sont beaucoup plus rares au diagnostic et surviennent plutôt en cours de traitement.
Infections
Les plus fréquentes concernent la sphère ORL, sinusienne ou bronchopulmonaire et sont le plus souvent à pneumocoque (Streptococcus pneumoniae) ou Haemophilus (Haemophilus influenzae) au début de la maladie, témoignant d’un déficit immunitaire humoral. En cours de traitement, apparaissent des infections à staphylocoques ou virales (réactivation d’herpès et de zona), voire opportunistes (pneumocystose à Pneumocystis jirovecii) en rapport avec un déficit immunitaire cellulaire favorisé par certains traitements. Il faut les prévenir par les vaccinations systématiques antigrippale, anti-Covid- 19 et antipneumococcique avant, et par une antibioprophylaxie, dès le début du traitement du myélome.
Hyperviscosité
Elle est rare au cours du myélome, contrairement à la maladie de Waldenström où l’IgM est pentamérique, et survient pour une protidémie supérieure à 130 g/L. Elle se manifeste par des épistaxis, des céphalées et des troubles de la vigilance, avec des exsudats et un courant granuleux au fond d’œil. Elle peut nécessiter la réalisation d’échanges plasmatiques avant le début en urgence du traitement spécifique.
Cryoglobulinémie
De type I, elle est exceptionnelle et liée à la précipitation de l’immunoglobuline monoclonale au froid (< 37 °C). Elle se manifeste par un purpura vasculaire cutané ou une glomérulonéphrite et peut rendre difficile l’évaluation quantitative du pic monoclonal si celle-ci n’est pas réalisée à 37 °C.
Principes du traitement
Traitement antitumoral
Indication
Seuls les patients symptomatiques, avec un critère CRAB ou SLIM-CRAB, sont traités. Les myélomes indolents font l’objet d’une surveillance clinique et biologique régulière pour dépister une progression vers un myélome symptomatique.
Médicaments
Les médicaments actifs au cours du myélome sont :
- les corticoïdes (prednisone et dexaméthasone) ;
- les alkylants (melphalan [Alkeran] et cyclophosphamide [Endoxan]) ;
- les inhibiteurs du protéasome, dont le chef de file est le bortézomib (Velcade) et s’administre par voie sous-cutanée, le carfilzomib étant administré par voie intraveineuse ;
- les immunomodulateurs (IMiD) : thalidomide, lénalidomide (Revlimid) et pomalidomide (Imnovid).
Indications thérapeutiques dépendant de l’âge du patient
Chez les sujets jeunes (moins de 66 ans), le traitement de référence est le traitement intensif : après un traitement d’induction, qui vise à réduire la masse tumorale et comporte une quadruple association corticoïdes-inhibiteur du protéasome-IMiD et anticorps monoclonal anti-CD38 (daratumumab ou isatuximab), le patient reçoit une chimiothérapie intensive par melphalan à forte dose et une réinjection de cellules souches hématopoïétiques autologues. Ce traitement peut être complété par une consolidation réutilisant l’association du traitement d’induction. Un traitement d'entretien par lénalidomide seul pendant 3 ans est actuellement validé.
Chez les sujets âgés de 66 ans et plus, ou présentant une contre-indication au traitement intensif, un traitement conventionnel est utilisé, associant les mêmes familles de médicaments : soit association melphalan-prednisone-bortézomib, soit, de façon plus récente, lénalidomide-dexaméthasone et daratumumab le plus souvent.
Traitement symptomatique
Il est essentiel.
L'anémie est traitée par transfusion et éventuellement érythropoïétine recombinante, en surveillant de près la correction du taux d’hémoglobine, pour interrompre le traitement à potentiel thrombogène.
Les infections sont traitées par une antibiothérapie précoce, adaptée et probabiliste (germes encapsulés) par amoxicilline ou équivalent, en évitant les médicaments néphrotoxiques. Le traitement substitutif par immunoglobulines parentérales peut être discuté en cas d’infections répétées. La vaccination antigrippale n’est pas contre-indiquée et est conseillée pour l’entourage proche du patient. Les vaccinations antipneumococcique et anti-Covid- 19 sont également conseillées en cas de myélome indolent ou en rémission et si possible avant de débuter le traitement spécifique.
Le traitement de l’insuffisance rénale (IR) repose sur la réhydratation pour corriger le facteur fonctionnel, la correction des troubles métaboliques associés (hyperuricémie, hypercalcémie), la dexaméthasone et, si besoin, l’épuration extrarénale. L'IR doit faire instaurer en urgence un traitement par bortézomib et dexaméthasone. Elle doit être prévenue par une bonne hydratation et l’éviction des facteurs favorisants, et notamment des médicaments néphrotoxiques.
Le traitement de l’hypercalcémie repose sur l’hydratation, la dexaméthasone et les bisphophonates par voie parentérale (zolédronate).
Le traitement de l’ostéopathie du myélome associe le traitement spécifique de la maladie, qui est rapidement antalgique, les antalgiques, y compris morphiniques, et les bisphosphonates. Les perfusions de bisphophonates, contemporaines des cycles de chimiothérapie, préviennent la survenue d’événements osseux en diminuant l’activation ostéoclastique. L’ostéonécrose aseptique de la mâchoire est une complication rare survenant lors de leur administration au long cours et doit être prévenue par une hygiène dentaire rigoureuse et l’éviction de tout foyer infectieux. La radiothérapie peut être indiquée pour un traitement antalgique rapide, de même que les traitements percutanés des tassements vertébraux (cimentoplastie ou kyphoplastie). Une intervention orthopédique préventive peut être nécessaire en cas de risque fracturaire.
Les urgences neurochirurgicales sont traitées par dexaméthasone puis radiothérapie ou chirurgie (laminectomie, vertébrectomie), suivie de radiothérapie, en fonction de la cause de la compression : épidurite isolée ou compression osseuse, par recul du mur vertébral postérieur, après avis neurochirurgical.
Évolution du myélome
La surveillance d’un myélome asymptomatique est régulière (tous les 3 à 6 mois) et comporte un examen clinique et un contrôle biologique de l’hémogramme, de la calcémie, de la créatininémie, de l’électrophorèse des protides sériques et de la protéinurie.
Le patient traité est évalué à chaque début de cycle pour la réponse au traitement et le dépistage de complications éventuelles ou d’effets indésirables liés au traitement. La réponse au traitement est suivie sur le taux de l’immunoglobuline monoclonale à l’électrophorèse des protides sériques et urinaires. Seuls les myélomes à chaînes légères et les exceptionnels myélomes non sécrétants justifient un dosage répété des chaînes légères libres sériques. La rémission peut être partielle ou complète, avec disparition de la plasmocytose médullaire. Malgré les progrès thérapeutiques qui ont permis un allongement significatif de la survie (dix ans en médiane chez les sujets jeunes), la guérison n’est pas encore d’actualité dans le myélome multiple.
Le diagnostic de myélome repose sur l’association d’une gammapathie monoclonale, complète ou non, et d’une plasmocytose médullaire d’au moins 10 %.
La gammapathie monoclonale est détectée par l’électrophorèse des protides sériques et urinaires pratiquée sur les urines des 24 heures. Ces examens permettent d’évaluer son taux et de le suivre sous traitement. L’immunofixation ou l’immunoélectrophorèse des protides sériques identifie l’immunoglobuline monoclonale (non IgM) mais ne permet pas sa quantification.
Le dosage des chaînes légères sériques n’est indiqué que pour les myélomes à chaînes légères ou « non sécrétants » suspectés devant une hypogammaglobulinémie à l’électrophorèse des protides sériques.
La maladie osseuse du myélome est due à un découplage du remodelage en faveur de l’ostéolyse qui est bloquée par l’administration de bisphosphonates.
L’imagerie moderne du myélome comporte un scanner du corps entier faible dose ou une IRM du corps entier.
La scintigraphie osseuse ne détecte pas les lésions du myélome (absence d’ostéoblastose).
Les urgences thérapeutiques du myélome comprennent l’hypercalcémie, la compression médullaire et la tubulopathie myélomateuse qui doit être prévenue.
Seuls les myélomes symptomatiques (critères CRAB) justifient un traitement spécifique qui inclut un traitement intensif avec autogreffe de cellules souches hématopoïétiques chez les patients jeunes.
Testez-vous sur cet item
Découvrez quelques quiz rédigés par les auteurs de cet item :
Rajkumar SV. Multiple myeloma: 2022 update on diagnosis, risk-stratification and management. Am J Hematol 2022;97:1086-107.
Vincent L, Roussel M, Macro M, Karlin L, Vekemans MC, Royer B, et al. Recommandations 2024 de l’Intergroupe francophone du myélome sur la prise en charge des gammapathies monoclonales de signification indéterminée. Hématologie 2024;30(1):71-92.
Vignon M, Bridoux F, Fermand JPP. Manifestations associées aux gammapathies monoclonales. Rev Prat 2018;68(7):792-6.
Fernandez de Larrea C et al. Blood Cancer Journal (2021) 11:192 ; https://doi.org/10.1038/s41408-021-00587-0
 Encadrés
Encadrés