La neurofibromatose de type 1 (NF1), ou maladie de von Recklinghausen, est une génodermatose prédisposant au développement de tumeurs, qu’elles soient bénignes ou malignes (encadré). Ces dernières années ont été marquées par des avancées significatives dans son diagnostic, son suivi et sa prise en charge. En France, la prise en charge de la NF1 s’organise en réseau national via le Centre de référence des neurofibromatoses (CERENEF, www.cerenef.org).
Mise à jour des critères diagnostiques de la NF1 en 2021
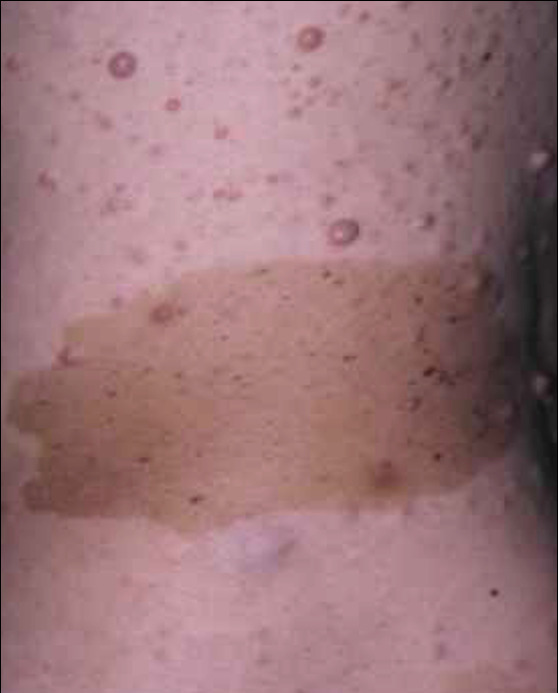
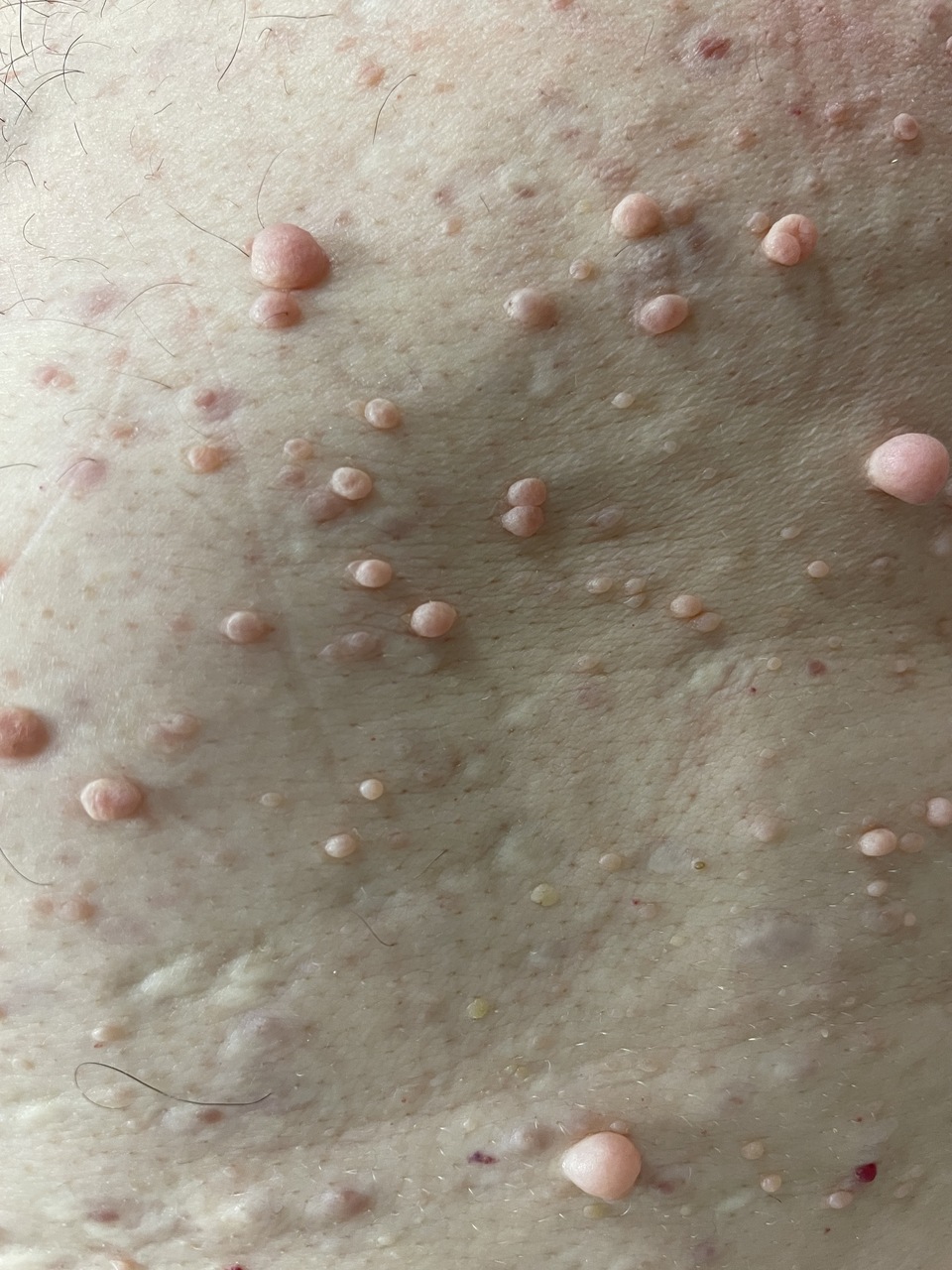
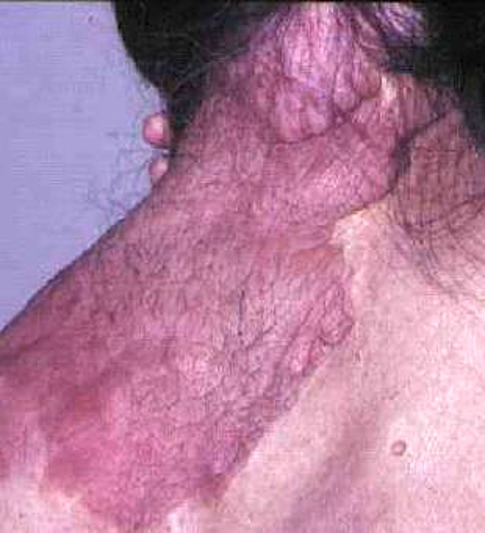
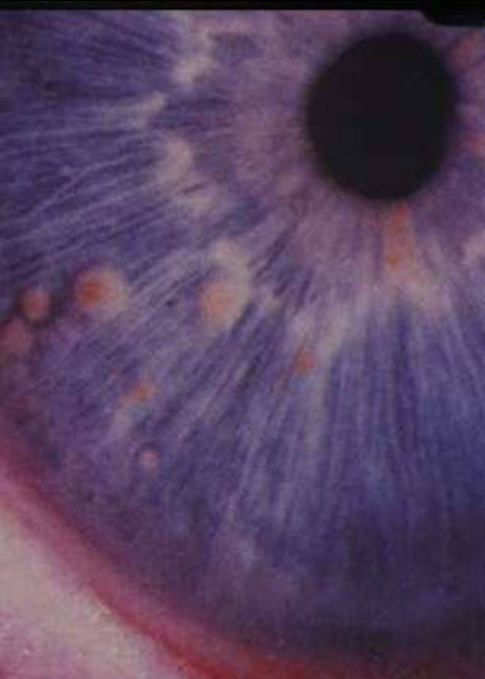
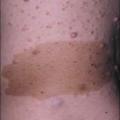
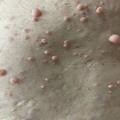
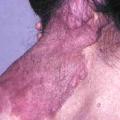
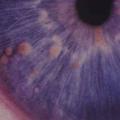
Les premiers critères diagnostiques de NF1 ont été définis en 1987, lors de la conférence de consensus dédiée aux neurofibromatoses des National Institutes of Health (NIH).1 Ceux-ci se fondaient principalement sur la clinique : taches café au lait (TCL) (fig. 1), lentigines, neurofibromes (fig. 2 et 3), nodules de Lisch (fig. 4), gliome optique et complications orthopédiques. Depuis cette date, les connaissances sur la symptomatologie de la NF1 se sont affinées et les outils d’analyse moléculaire se sont développés ; ces avancées permettent aujourd’hui de détecter une mutation du gène causal, NF1, dans 95 % des cas.2
En 2021, un groupe international d’experts des neurofibromatoses et schwannomatoses constitué en consortium s’est réuni pour réviser les critères diagnostiques de la NF1, de la NF2 et des schwannomatoses. Pour la NF1, les principales modifications concernent les cinq éléments suivants :
- le caractère obligatoirement bilatéral d’au moins une manifestation pigmentaire (TCL ou lentigines) ;
- l’introduction des hamartomes choroïdiens (ou taches de Yasunari) à l’examen ophtalmologique ;
- la définition plus précise des anomalies orthopédiques (courbure antérolatérale de tibia, pseudarthrose d’un os long) ;
- l’introduction de l’analyse moléculaire (présence d’un variant hétérozygote du gène NF1, avec une fraction allélique de 50 %) ;
- des modifications sémantiques (tableau).3
Ce travail a également permis de préciser les critères diagnostiques du principal diagnostic différentiel de la NF1, que constitue le syndrome de Legius. Dans ce dernier, également caractérisé par des anomalies pigmentaires (TCL, lentigines), aucun neurofibrome ne se développe et le risque de transformation sarcomateuse n’existe pas.
Ont également été définis les critères diagnostiques des formes dites mosaïques ou segmentaires de NF1. Celles-ci, conséquences d’une mutation, non pas constitutionnelle mais post-zygotique, sont classiquement peu sévères et de topographie souvent localisée.3,4
Dépistage précoce du cancer du sein chez la jeune femme atteinte de NF1
La NF1 est associée à un sur-risque (multiplié par 2 à 3) de développer un cancer.5 Le cancer du sein chez les femmes atteintes de NF1 se caractérise par une survenue plus précoce (avant l’âge de 50 ans) et une mortalité plus importante (jusqu’à 5 fois supérieure) .6 Le risque de développer ce cancer (de sous-type majoritaire : carcinome intracanalaire infiltrant) est estimé entre 2 et 5 fois plus élevé que dans la population générale,7 et le risque cumulé de développer un cancer controlatéral est de 27 %, à vingt ans du premier diagnostic.8
Ainsi considérées comme des personnes à « risque très élevé » de cancer du sein, les femmes atteintes de NF1 doivent être dépistées précocement. Ces modalités de dépistage ont été détaillées dans le Protocole national de diagnostic et de soins (PNDS) de la NF1 publié en 2021 :9 dès l’âge de 30 ans, il est recommandé de réaliser de manière annuelle une imagerie par résonance magnétique (IRM) mammaire et une mammographie (avec une seule incidence : oblique externe), éventuellement complétées par une échographie. Dans cette population, la balance bénéfices-risques de l’irradiation est en faveur du dépistage précoce. Toutefois, l’interprétation de ces examens est parfois difficile, du fait d’éventuels neurofibromes cutanés et/ou intra-mammaires.
Surveillance des patients à risque de développer une tumeur maligne des gaines nerveuses
Les tumeurs malignes des gaines nerveuses (TMGN) représentent le cancer le plus redouté chez les individus atteints de NF1, touchant en particulier le jeune adulte, et cause essentielle de leur mortalité.10
Deux principaux facteurs de risque de développer une TMGN ont été mis en évidence ; la présence de l’un des deux nécessite une vigilance particulière :
- dix neurofibromes sous-cutanés ou plus ;
- au moins un neurofibrome interne.11 Il n’est toutefois pas recommandé de rechercher de manière systématique, par imagerie, un neurofibrome interne mais seulement en cas de point d’appel clinique (apparition ou augmentation de volume d’une masse, douleur, déficit neurologique, etc.).
En pratique, dans cette population à risque (contexte de dépistage ciblé) et/ou si la clinique fait suspecter une transformation maligne (contexte de diagnostic devant l’un des points d’appel sus-décrits et/ou devant une altération de l’état général), les experts recommandent la réalisation d’une IRM corps entier couplée à une tomodensitométrie par émission de positons (TEP) [TEP-TDM ou TEP-IRM] initiales.
Le dépistage ciblé (par TEP-IRM), dans les limites de sa disponibilité, a récemment démontré sa capacité à diagnostiquer précocement des TMGN asymptomatiques.12 La fréquence de réalisation des imageries ultérieures dépend des résultats des investigations initiales et fait régulièrement l’objet de discussions en réunions de concertation pluri-disciplinaires (RCP) locales et/ou nationales.
Traitement des neurofibromes plexiformes inopérables de l’enfant par inhibiteur de MEK
Dans la NF1, la voie RAS est suractivée de manière continue, intégrant, de ce fait, cette pathologie dans le groupe des RASopathies. Depuis une décennie, la meilleure compréhension du rôle de cette voie de signalisation (RAS/RAF/MEK/ERK) dans la prolifération cellulaire a orienté les recherches vers l’étude des inhibiteurs de MEK dans la NF1 autour de plusieurs de ses complications.
En 2016, l’efficacité du sélumétinib sur les neurofibromes plexiformes inopérables (NFPI) de l’enfant a été mise en évidence dans une étude de phase I ;13 ces résultats ont été confirmés en 2020 dans l’étude de phase II SPRINT : 37 enfants sur 50 (74 % ; intervalle de confiance [IC] à 95 % : 60 - 85) ont présenté une réponse partielle (diminution de volume ≥ 20 %), confirmée à trois mois chez 35 sur 50 (70 %) et durable (maintenue sur ≥ 12 cycles) chez 28 sur 50 (56 %). À ces résultats volumétriques se sont ajoutées des données témoignant de l’amélioration de la qualité de vie (PedsQL), de l’activité physique (PROMIS), de la douleur (NRS- 11) et de la « déformation inesthétique » (échelle d’impression globale de changement, échelle de Likert notée sur 7).
Les effets indésirables les plus fréquemment rapportés sont de grades 1 et 2 : symptômes digestifs (nausées, vomissements, diarrhées, n = 38/50 ; 76 %), élévation asymptomatique des créatines phosphokinases [CPK] (n = 36/50 ; 72 %), éruption acnéiforme (n = 27/50 ; 54 %) et paronychie (n = 18/50 ; 38 %).
Aucun effet indésirable grave notamment cardiaque ou ophtalmologique n’a été observé. Pour 14 patients (28 %), une réduction de dose a été nécessaire du fait d’effets indésirables. La posologie de sélumétinib retenue est de 25 mg/m2 toutes les douze heures.14
Ces travaux ont permis, depuis 2020 et pour la première fois, qu’un traitement soit approuvé dans la NF1, par la Food Drug Administration (FDA), puis par l’Agence européenne des médicaments (EMA) en juin 2021. En France, le sélumétinib est commercialisé depuis 2022 dans l’indication de traitement du NFPI symptomatique chez l’enfant atteint de NF1 à partir de l’âge de 3 ans. Il peut être prescrit sur ordonnance simple, par des spécialistes hospitaliers compétents.
Le sélumétinib est actuellement en cours d’évaluation chez l’adulte dans la même indication.
Recherches à poursuivre
Ces dernières années, la NF1 a fait l’objet d’importants travaux de recherche ayant permis des avancées significatives dans le diagnostic, le dépistage (généralisé ou ciblé) et la prise en charge des personnes qui en sont atteintes. Les nombreux projets actuels et futurs nourrissent l’espoir de nouvelles thérapeutiques ciblant les neurofibromes, qu’ils soient plexiformes ou cutanés, et leur transformation maligne.
Qu’est-ce que la Neurofibromatose de type 1 ?
La neurofibromatose de type 1 (NF1), ou maladie de von Recklinghausen, est une maladie génétique de transmission autosomique dominante. Elle touche environ 1 naissance sur 3 000, soit environ 20 000 personnes, en France. Cette pathologie prédispose au développement de tumeurs, qu’elles soient bénignes ou malignes. Les neurofibromes cutanés sont les tumeurs bénignes les plus fréquemment rencontrées chez les individus atteints de NF1, touchant plus de 95 % d’entre eux. Le diagnostic repose sur des critères majoritairement cliniques révisés en 2021. La pénétrance de la NF1 est complète dès l’âge de 8 ans. La prise en charge et le suivi des patients, enfants ou adultes, nécessitent un abord multidisciplinaire. En France, elle est coordonnée par le Centre de référence des neurofibromatoses (www.cerenef.org).
2. Messiaen LM, Callens T, Mortier G, et al. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum Mutat 2000;15:541–55.
3. Legius E, Messiaen L, Wolkenstein P, et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet Med 2021:23(8):1506-13.
4. Fertitta L, Bergqvist C, Matthieu R, et al. Version française des critères révisés des diagnostics de neurofibromatose de type 1, de syndrome de Legius et de leurs formes mosaïques. Ann Dermatol Venereol 2022;2(6):464-8.
5. Seminog OO, Goldacre MJ. Risk of benign tumours of nervous system, and of malignant neoplasms, in people with neurofibromatosis: Population-based record-linkage study. Br J Cancer 2013;108:193-8.
6. Uusitalo E, Kallionpää RA, Kurki S, et al. Breast cancer in neurofibromatosis type 1: Overrepresentation of unfavourable prognostic factors. Br J Cancer 2017;116:211-7.
7. Breast Cancer Association Consortium, Dorling L, Carvalho S, et al. Breast Cancer Risk Genes - Association analysis in more than 113,000 women. N Engl J Med 2021;384:428-39.
8. Evans DGR, Kallionpää RA, Clementi M, et al. Breast cancer in neurofibromatosis 1: Survival and risk of contralateral breast cancer in a five country cohort study. Genet Med 2020;22(2):398-406. 9. Protocole national de diagnostic et de soins (PNDS) Neurofibromatose 1. 2021. https://www.has-sante.fr/upload/docs/application/pdf/2022-07/pndsnf1final.pdf
10. Evans DGR, Baser ME, McGaughran J, et al. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet 2002;39:311-4.
11. Sbidian E, Bastuji-Garin S, Valeyrie-Allanore L, et al. At-risk phenotype of neurofibromatose-1 patients: A multicentre case-control study. Orphanet J Rare Dis 2011;6:51.
12. Fertitta L, Jannic A, Zehou O, et al. Whole-body 18F-FDG-PET/MRI as a screening tool for the detection of malignant transformation in individuals with neurofibromatosis type 1. J Invest Dermatol 2024:S0022-202X(24)00112-X.
13. Dombi E, Baldwin A, Marcus LJ, et al. Activity of selumetinib in neurofibromatosis type 1-related plexiform neurofibromas. N Engl J Med 2016;375:2550-60.
14. Gross AM, Wolters PL, Dombi E, et al. Selumetinib in children with inoperable plexiform neurofibromas. N Engl J Med 2020;382:1430-42.
Dans cet article
- Mise à jour des critères diagnostiques de la NF1 en 2021
- Dépistage précoce du cancer du sein chez la jeune femme atteinte de NF1
- Surveillance des patients à risque de développer une tumeur maligne des gaines nerveuses
- Traitement des neurofibromes plexiformes inopérables de l’enfant par inhibiteur de MEK
- Recherches à poursuivre
 Encadrés
Encadrés