La mucoviscidose est la maladie génétique sévère la plus fréquente dans la population caucasienne. Un individu sur 35 est porteur d’un variant du gène CFTR à l’état hétérozygote, et la mucoviscidose atteint 1 nouveau-né sur 5 000. Depuis 2002, le dépistage néonatal est systématiquement proposé aux parents ; il existe néanmoins des patients encore non diagnostiqués : faux négatifs du dépistage néonatal, nés avant 2002 ou dans un pays étranger ne pratiquant pas le dépistage néonatal. Le pronostic des patients atteints de mucoviscidose a été bouleversé par l’arrivée en France, début 2020, d’une triple association de molécules modulatrices de la protéine CFTR (élexacaftor-tézacaftor-ivacaftor [ETI]).
Mucoviscidose : qu’est-ce que c’est ?
La mucoviscidose est une maladie génétique autosomique récessive causée par des mutations du gène CFTR, porté par le chromosome 7, codant pour une protéine exprimée à la membrane apicale de nombreux épithéliums du corps humain. La protéine CFTR est un canal ionique qui excrète principalement les ions chlorure et bicarbonate depuis l’intérieur de la cellule vers le milieu extracellulaire (fig. 1). La sortie d’ions chlorure chargés négativement entraîne une sortie d’ions sodium qui attire l’eau vers le milieu extérieur. La protéine CFTR a donc pour principale fonction d’hydrater le liquide péricellulaire et d’assurer une bonne fonctionnalité du mucus sécrété par les épithéliums, notamment respiratoire, mais aussi dans d’autres organes comme les sinus, la peau, le foie, le pancréas, le tube digestif et le tractus génital. La mucoviscidose, dont le pronostic est lié à l’atteinte pulmonaire, est responsable de nombreuses manifestations extra-respiratoires, de date de survenue, fréquence et intensité variables :
- déshydratation aiguë/coups de chaleur du fait de l’hypersudation ;
- obstruction nasale chronique sur polypose nasosinusienne et sinusites à répétition ;
- pancréatites aiguës récidivantes ;
- stéatorrhée et malabsorption sur insuffisance pancréatique exocrine ;
- diabète ;
- infertilité masculine sur agénésie des canaux déférents ;
- cirrhose et hypertension portale ;
- iléus méconial ;
- syndrome d’occlusion intestinale distal ;
- troubles digestifs chroniques ;
- dermatose palmoplantaire aquagénique.
À l’heure actuelle, plus de 2 100 mutations du gène CFTR sont identifiées, avec des conséquences cliniques variables. Elles sont définies depuis plusieurs dizaines d’années selon leur appartenance à l’une ou l’autre des six classes existantes (fig. 1). L’élément le plus important à retenir et qui retentit sur la probabilité de réponse ou non au traitement par ETI, modulateur de CFTR, est la synthèse ou non de la protéine CFTR.
De la symptomatologie respiratoire dépend le pronostic des patients. Du fait d’une clairance mucociliaire altérée, leurs bronches sont infectées dès le plus jeune âge, avec des symptômes tels que bronchite chronique, toux et expectorations épaisses et collantes. Au gré des surinfections bronchiques se développent des lésions à type d’inflammation de la paroi et d’obstruction bronchique par un mucus épais, conduisant au développement progressif de dilatations de bronches diffuses.
L’évolution, en l’absence de traitement, se fait souvent vers l’insuffisance respiratoire chronique, qui peut conduire à la transplantation pulmonaire ou au décès, avec parfois des complications aiguës et sévères telles que l’aspergillose bronchopulmonaire allergique, les hémoptysies ou les pneumothorax spontanés secondaires.
Modulateurs de CFTR : quand et pour qui ?
Des traitements d’abord uniquement symptomatiques
Jusqu’en 2011, l’arsenal thérapeutique de la mucoviscidose ne comportait que des traitements principalement symptomatiques, visant à corriger :
- l’insuffisance pancréatique exocrine, par la supplémentation en enzymes pancréatiques, en vitamines liposolubles et l’instauration d’un régime hypercalorique et riche en graisses ;
- l’infection bronchique chronique, par des antibiothérapies adaptées à la flore du patient (antibiothérapies orales alternées systématiques, intraveineuses prolongées, ou inhalées séquentielles) ;
- l’obstruction bronchique, par l’amélioration de la clairance mucociliaire (aérosol-thérapie de sérum salé hypertonique ou de dornase alfa, kinésithérapie respiratoire) et la bronchodilatation par bêta- 2 -mimétiques et anticholinergiques de courte et longue durées d’action, tout en évitant la prescription non justifiée de corticoïdes inhalés ;
- le diabète, par un suivi endocrinologique spécialisé ;
- l’infertilité, par une prise en charge en centre d’assistance médicale à la procréation.
Ivacaftor, premier des odulateurs de CFTR
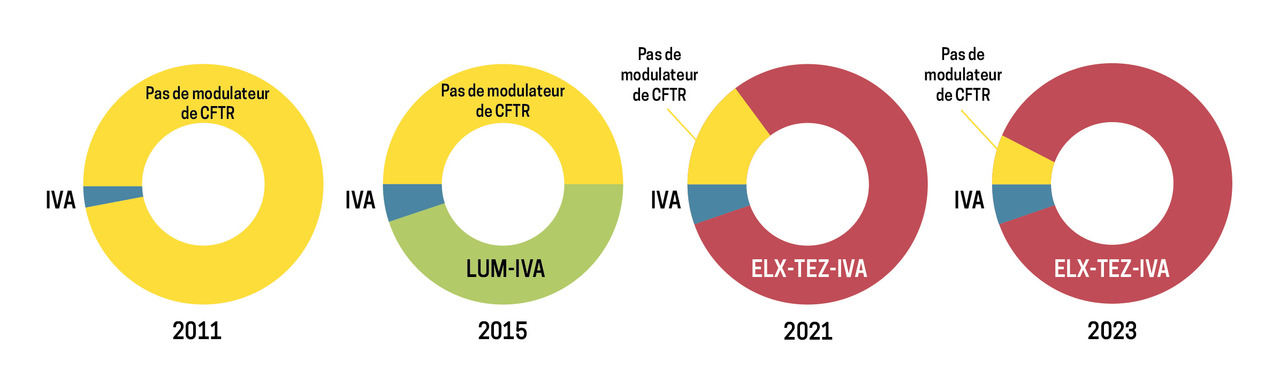
En 2011, le premier traitement modulateur de CFTR est apparu : l’ivacaftor (Kalydeco). Il n’était, à l’époque, indiqué que pour les patients porteurs d’une mutation G551D (classe 3) ; l’autorisation de mise sur le marché (AMM) en France a été obtenue en 2013.
Les études ont montré, pour la première fois dans l’histoire de la mucoviscidose, qu’un traitement pouvait induire une amélioration significative du volume expiratoire maximal en une seconde (VEMS) [de 10 %] et de l’indice de masse corporelle (IMC), et une diminution des concentrations sudorales en ions chlorure.1
Plus récemment, ses indications ont été élargies à d’autres mutations, toutes de classe 3 (mutations avec synthèse de protéine exprimée à la membrane cellulaire, mais avec un défaut d’ouverture du canal ionique CFTR [fig. 1]).
L’ivacaftor est en effet une petite molécule, dite « correctrice » de CFTR, capable d’interagir avec la protéine CFTR et de stabiliser le canal en position ouverte, ce qui restaure le transport d’ions chlorure chez les patients porteurs de ce type de mutations.
L’ivacaftor a ensuite été testé sans succès chez les patients porteurs homozygotes de la mutation phe508del (mutation la plus fréquente dans la population caucasienne). Ce résultat négatif est expliqué par le fait que, dans les mutations de classe 2, comme la phe508del, une protéine CFTR est bien synthétisée mais n’est pas exprimée à la membrane apicale (fig. 1).
Première association de Modulateurs de CFTR : peu convaincante
En 2015, le même laboratoire a développé la première association de modulateurs de CFTR : ivacaftor-lumacaftor (Orkambi). Le lumacaftor est un « potentiateur » de CFTR qui interagit avec la protéine CFTR anormale et facilite son adressage et son expression à la membrane cellulaire. Une fois exprimée, la protéine peut alors interagir avec l’ivacaftor et ainsi récupérer une fonction canal.
Les premières études sur l’impact de l’association ivacaftor-lumacaftor chez les patients porteurs homozygotes de la mutation phe508del étaient positives, quoique décevantes : le gain de VEMS n’était que de 3 % et les autres paramètres, en dehors de l’IMC, n’étaient pas modifiés.2 Des études françaises en vraie vie ont confirmé l’efficacité modérée sur le VEMS et sur la fréquence des exacerbations respiratoires mais ont également mis en avant une fréquence importante d’effets indésirables, notamment respiratoires, d’autant plus marqués que la fonction respiratoire de base du patient était altérée. L’intensité de ces effets indésirables (bronchospasmes principalement) a conduit à l’interruption du traitement chez environ 18 % des patients à un an de suivi et jusqu’à 30 % des patients avec un VEMS inférieur à 40 %.3,4
Trithérapie modulatrice de CFTR, une révolution
En octobre 2019 ont été publiées les deux premières études décrivant l’efficacité de la trithérapie modulatrice élexacaftor-tézacaftor-ivacaftor (Kaftrio) chez des patients porteurs d’au moins une mutation phe508del. Les patients déjà traités par ivacaftor-lumacaftor ont eu une amélioration du VEMS de 10 % sous ETI ; ceux sans traitement modulateur de CFTR préalable ont eu un gain de 14 % en moyenne, avec également un gain significatif sur l’IMC, une diminution significative de la concentration en chlorures sudoraux, une augmentation significative du délai avant la prochaine exacerbation et, pour la première fois, une amélioration significative de la qualité de vie, avec disparition des symptômes respiratoires quotidiens.5 Ces données ont été confirmées en vraie vie par plusieurs études françaises, chez des patients plus sévères à l’initiation de l’ETI (VEMS inférieur à 40 % de la valeur prédite et, pour certains, en cours de projet de transplantation pulmonaire) qui, de surcroît, ont eu une amélioration significative de qualité de vie, une diminution des besoins en oxygène et en ventilation non invasive et, surtout, un changement de trajectoire avec, pour la très grande majorité, la disparition de l’indication de transplantation pulmonaire.6 - 8
Progressivement, depuis juillet 2021, l’ETI est devenu accessible en France à tous les patients atteints de mucoviscidose porteurs d’au moins une mutation phe508del âgés de 6 ans et plus, quel que soit l’état de leur fonction respiratoire.9 Une extension d’AMM a été demandée pour les patients de 2 à 5 ans. Celle-ci a abouti fin 2023. La Food and Drug Administration (FDA) a secondairement approuvé 177 mutations supplémentaires comme étant éligibles à l’ETI, sur critères contestables et exclusivement in vitro. En conséquence, ni l’Agence européenne des médicaments (EMA) ni les autorités françaises du médicament n’ont modifié les conditions de prescription de l’ETI à la suite de la prise de position de la FDA (fig. 2).
Perspectives des modulateurs de CFTR : l’exception française
En mai 2022, en collaboration avec l’Agence nationale de sécurité du médicament (ANSM) et l’association Vaincre la mucoviscidose, le Centre national de référence de mucoviscidose a mis au point un programme compassionnel de prescription de l’ETI pour les patients atteints de la mucoviscidose âgés de 12 ans et plus ayant une forme sévère (VEMS inférieur à 40 % de la valeur prédite) et non porteurs de la mutation phe508del. Les patients remplissant ces critères ont pu bénéficier de deux mois de traitement, avec une évaluation après quatre à six semaines. La réponse clinique a été évaluée sur des critères cliniques, spirométriques, radiologiques et physiologiques (amélioration du test de la sueur). Après évaluation par le Centre national de référence, le traitement était renouvelé pour le patient considéré comme répondeur ; dans le cas contraire, il était interrompu.10 Près de 84 patients ont été inclus dans le programme. Après quatre à six semaines de traitement, 45 patients ont été considérés comme répondeurs. Parmi eux, 22 étaient porteurs de mutations non approuvées par la FDA. Tous les patients répondeurs ont rapporté une diminution, voire une disparition, des symptômes ; 1 patient a été retiré de liste de transplantation et seuls 2 parmi les 11 en cours d’inscription sur la liste de greffe au moment de débuter le traitement restaient encore en voie d’inscription. Au vu des résultats de cette étude en vraie vie sur l’efficacité de l’ETI chez des patients sévères non porteurs de la mutation phe508del, l’ANSM a publié, le 1er juin 2023, l’extension officielle du programme aux patients atteints de mucoviscidose âgés de 6 ans et plus non porteurs de mutations phe508del, quel que soit l’état de leur fonction respiratoire.
Ainsi, grâce à ce programme, l’accès au traitement par ETI est élargi à tous les patients atteints de mucoviscidose, avec une évaluation de l’efficacité en vraie vie. Avec l’AMM actuelle, 85 à 90 % des patients sont traités ; avec le programme compassionnel, la proportion de patients avec un bénéfice clinique prouvé de l’ETI pourrait augmenter. Au fur et à mesure des évaluations, certaines mutations, notamment celles de classe 1 incluant des codons stop, sont néanmoins écartées, car définies avec certitude comme non répondeuses.
La mucoviscidose est une pathologie sévère dont le pronostic a été transformé pour la très grande majorité des patients ; il reste toutefois une minorité à ne pas oublier et pour laquelle d’autres pistes thérapeutiques sont en cours d’évaluation.
Que dire à vos patients ?
La mucoviscidose est une pathologie génétique sévère dont le pronostic a été transformé par l’arrivée du traitement associant trois molécules (élexacaftor-tézacaftor-ivacaftor) ; il est donc essentiel de se donner les moyens de diagnostiquer la mucoviscidose, y compris à l’âge adulte.
Les centres maladies rares-mucoviscidose sont répartis dans toute la France ; le site de l’association Vaincre la mucoviscidose les répertorie : https ://www.vaincrelamuco.org/pres-de-chez-vous#tab1.
Lorsqu’un diagnostic de mucoviscidose est posé, le patient et ses apparentés sont adressés en consultation de conseil génétique.
2. Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med 2015;373(3):220-31.
3. Burgel PR, Munck A, Durieu I, et al. Real-life safety and effectiveness of lumacaftor-ivacaftor in patients with cystic fibrosis. Am J Crit Care Med 2020;201(2):188-97.
4. Burgel PR, Durieu I, Chiron R, et al. Clinical response to lumacaftor-ivacaftor in patients with cystic fibrosis according to baseline lung function. J Cyst Fibros 2021;20(2):220-7.
5. Middleton PG, Mall MA, Dřevínek P, et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N Engl J Med 2019;381(19):1809-19.
6. Burgel PR, Durieu I, Chiron R, et al. Rapid Improvement after Starting Elexacaftor-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and Advanced Pulmonary Disease. Am J Respir Crit Care Med 2021;204(1):64-73.
7. Martin C, Reynaud-Gaubert M, Hamidfar R, et al. Sustained effectiveness of elexacaftor-tezacaftor-ivacaftor in lung transplant candidates with cystic fibrosis. J Cyst Fibros 2022;21(3):489-96.
8. Martin C, Burnet E, Ronayette-Preira A, et al. Patient perspectives following initiation of elexacaftor-tezacaftor-ivacaftor in people with cystic fibrosis and advanced lung disease. Respir Med Res 2021;80:100829.
9. Regard L, Martin C, Burnet E, et al. CFTR Modulators in People with Cystic Fibrosis: Real-World Evidence in France. Cells 2022;11(11):1769.
10. Burgel PR, Sermet-Gaudelus I, Durieu I, et al. The French Compassionate Program of elexacaftor-tezacaftor-ivacaftor in people with cystic fibrosis with advanced lung disease and no F508del CFTR variant. Eur Respir J 2023:2202437.
Dans cet article
 Encadrés
Encadrés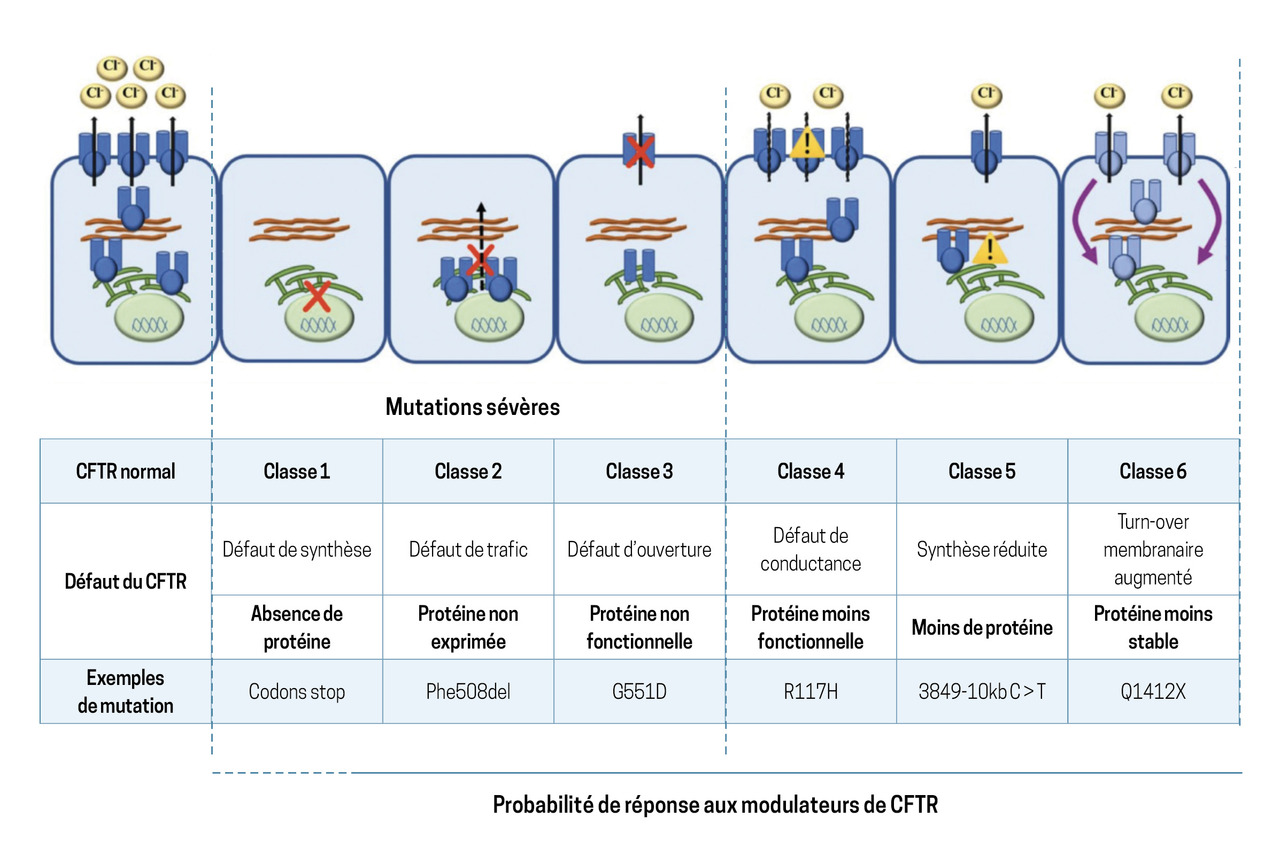
Les patients avec dilatation des bronches diffuse doivent être adressés en pneumologie pour bénéficier d’une enquête étiologique complète. À la moindre suspicion de mucoviscidose (atteinte sinusienne et/ou digestive, survenue des symptômes depuis l’enfance), un avis auprès d’un centre maladies rares-mucoviscidose doit être pris.
Les progrès thérapeutiques importants des cinq dernières années justifient la recherche large du diagnostic de mucoviscidose chez les patients atteints de dilatations des bronches, du fait d’un bénéfice individuel majeur de ces traitements.
Chez les patients répondeurs à la trithérapie modulatrice de la protéine CFTR (élexacaftor-tézacaftor-ivacaftor), la qualité de vie et le pronostic vital sont transformés.