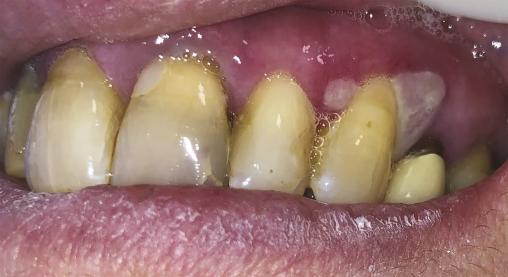
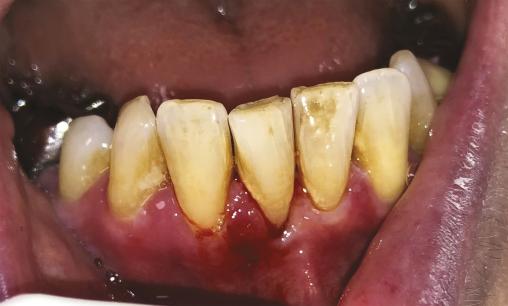
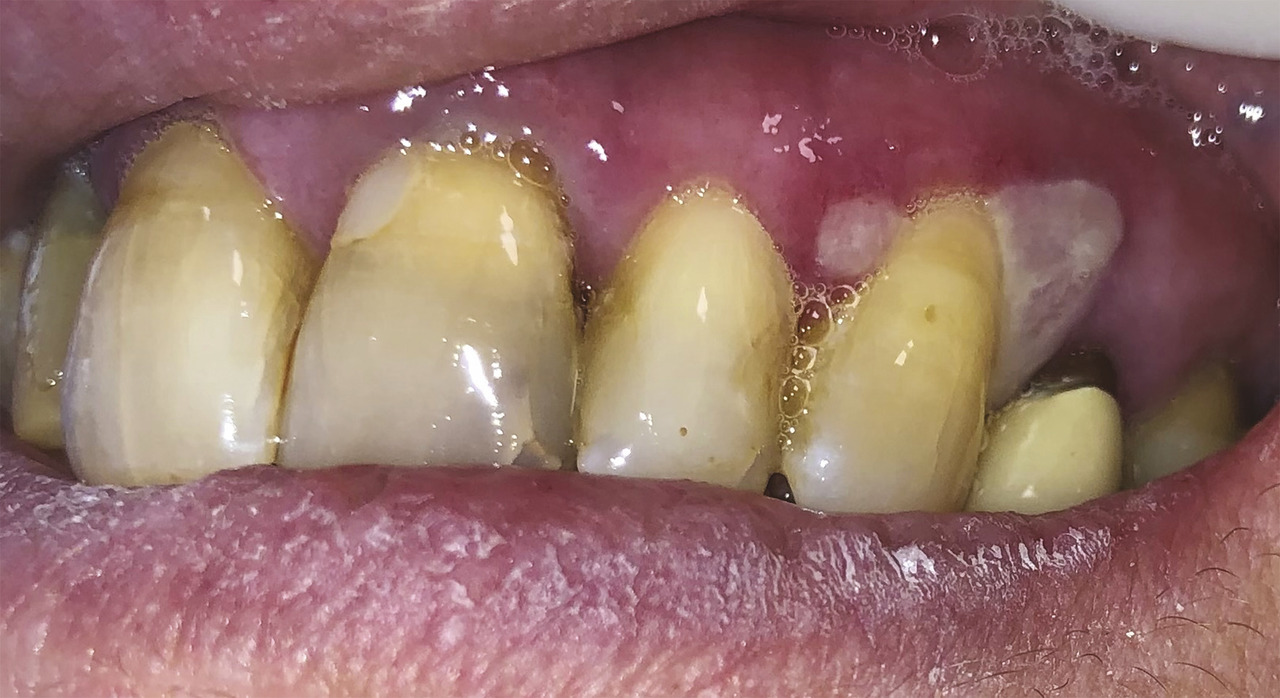
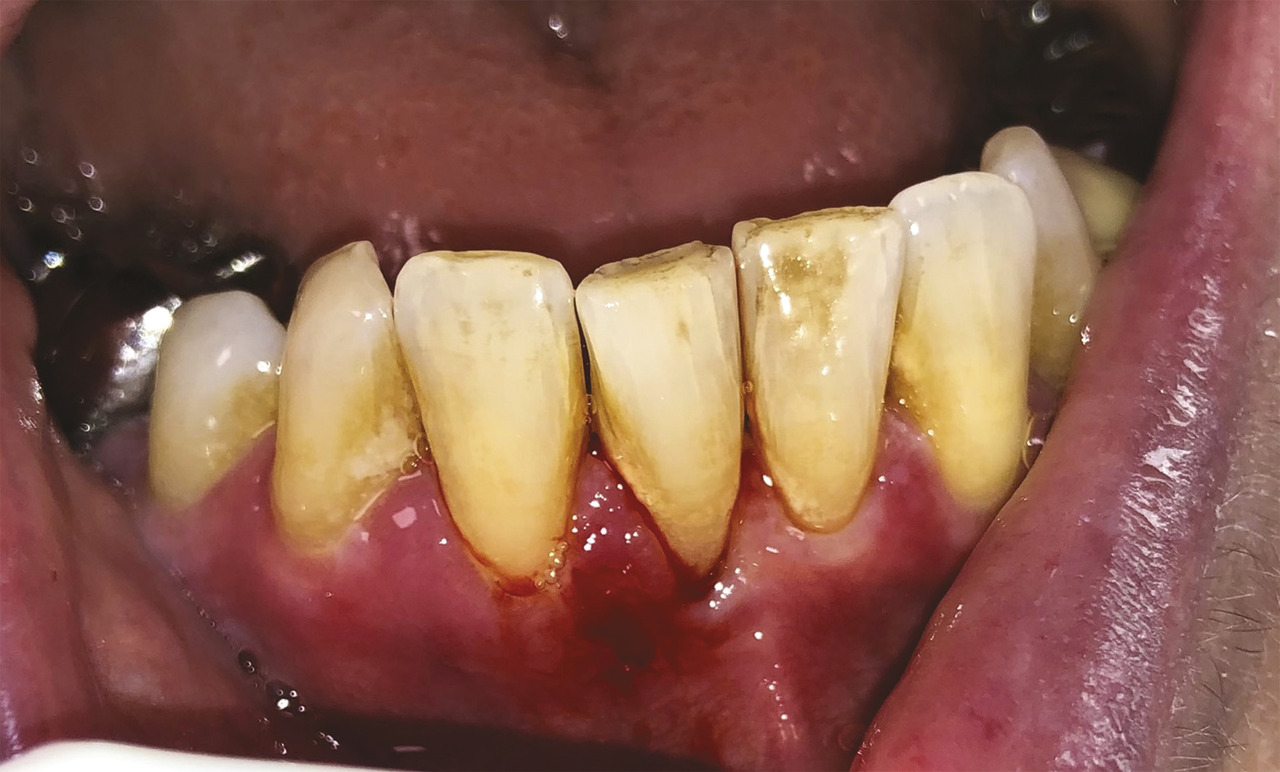
Une patiente de 61 ans consulte pour une gingivorragie diffuse persistante et douloureuse, ainsi que des ulcérations buccales récurrentes au cours des trois derniers mois. Ses antécédents médicaux comprennent une thyroïdectomie substituée par une hormonothérapie et un reflux gastro-œsophagien. On note l’absence d’intoxication alcoolo-tabagique. La patiente ne rapporte pas de modification récente de ses traitements, de son alimentation, de son dentifrice ou de ses habitudes de vie, ni de symptôme extra-oral. Malgré l’éviction des aliments acides tels que le citron ou le vinaigre, ses symptômes persistent. L’examen endobuccal révèle des lésions gingivales hyperkératosiques et érythémateuses, une lésion bulleuse gingivale (fig. 1 ) et plusieurs érosions (fig. 2 ) associées à une gingivite légère. Le signe de Nikolsky est positif, correspondant à l’érosion induite par un frottement sur une peau ou une muqueuse d’apparence saine, suggérant une maladie bulleuse. Aucune adénopathie cervicofaciale n’est objectivée lors de l’examen clinique.
Les analyses sanguines sont normales, permettant d’exclure les hypothèses de carences nutritionnelles, d’immunodépression et d’hémopathies malignes.
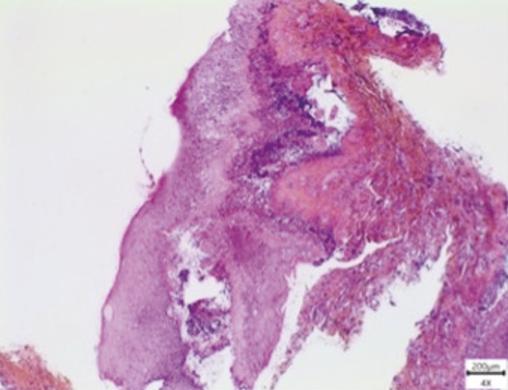
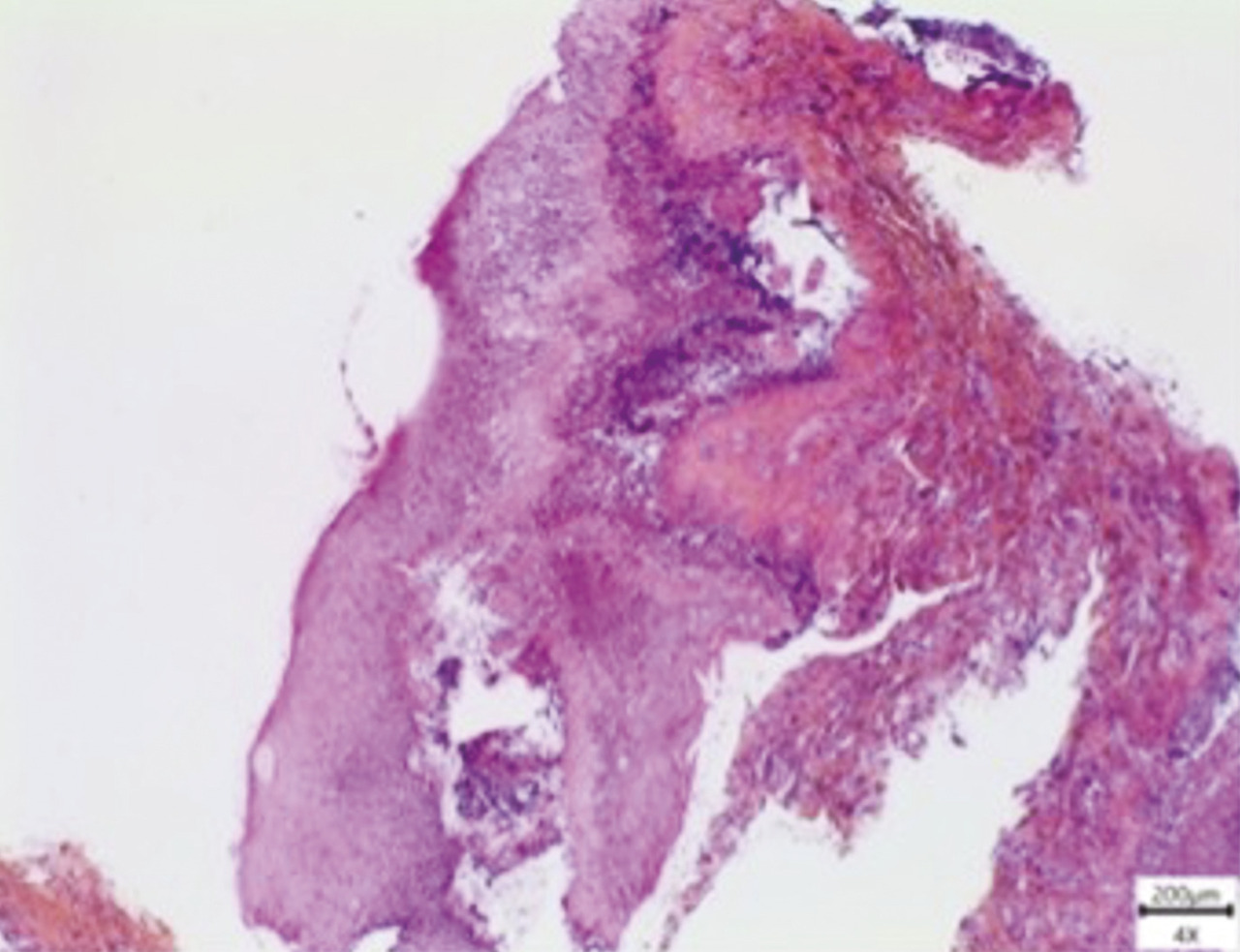
Une biopsie est effectuée, à cheval sur une lésion gingivale érosive et la muqueuse périlésionnelle. L’examen histologique révèle un toit bulleux composé d’épithélium, rempli de sérosités et d’un infiltrat inflammatoire abondant, principalement constitué de polynucléaires neutrophiles (fig. 3 ). Par la suite, une analyse en immunofluorescence directe met en évidence la présence de dépôts d’immunoglobulines G (IgG) et de fractions de complément (C3) le long de la membrane basale, cohérent avec le diagnostic de pemphigoïde bulleuse. Les dosages d’anticorps anti-BP180 et anti-BP230 sont négatifs.
Une corticothérapie locale probabiliste est initiée (prednisolone 20 mg en bain de bouche trois fois par jour durant trois semaines, puis diminution posologique progressive durant trois semaines). Des soins locaux par des bains de bouche à la chlorhexidine et au bicarbonate de sodium sont associés, ainsi que du paracétamol à visée antalgique. Les lésions ont totalement disparu en deux semaines. Un suivi régulier semestriel a été programmé.
Les analyses sanguines sont normales, permettant d’exclure les hypothèses de carences nutritionnelles, d’immunodépression et d’hémopathies malignes.
Une biopsie est effectuée, à cheval sur une lésion gingivale érosive et la muqueuse périlésionnelle. L’examen histologique révèle un toit bulleux composé d’épithélium, rempli de sérosités et d’un infiltrat inflammatoire abondant, principalement constitué de polynucléaires neutrophiles (
Une corticothérapie locale probabiliste est initiée (prednisolone 20 mg en bain de bouche trois fois par jour durant trois semaines, puis diminution posologique progressive durant trois semaines). Des soins locaux par des bains de bouche à la chlorhexidine et au bicarbonate de sodium sont associés, ainsi que du paracétamol à visée antalgique. Les lésions ont totalement disparu en deux semaines. Un suivi régulier semestriel a été programmé.
La pemphigoïde bulleuse est une maladie auto-immune chronique causée par des anticorps ciblant les protéines structurelles de la membrane basale épithéliale. Cliniquement, elle se caractérise par de larges bulles sous-épithéliales affectant les surfaces cutanées et/ou muqueuses.1 La muqueuse orale est impliquée dans 10 à 20 % des cas. Les signes sont des érythèmes, bulles, érosions ou ulcérations.2-5 La pemphigoïde bulleuse orale se présente fréquemment sous forme d’ulcérations ou de gingivites, l’observation directe de bulles étant rare.3,5 Les stades ultérieurs de la maladie impliquent la peau2 et provoquent des plaintes sévères telles que des douleurs, sensations de brûlure et dysphagie, compromettant significativement la qualité de vie.1,6
Le diagnostic repose sur l’examen histologique d’un échantillon de bulle frais, objectivant un clivage sous-épidermique associé à un infiltrat inflammatoire.5,7 L’immunofluorescence directe est la technique de référence, montrant un dépôt linéaire d’immunoglobulines G (IgG), A (IgA) ou de fractions du complément (C3) le long de la membrane basale épithéliale.1,2,4,8-10 Des tests immunosérologiques (test ELISA mesurant les taux sériques d’anticorps dirigés contre les protéines BP180 et BP230) sont couramment employés comme outils diagnostiques.1,2,8,10 Cependant, les données de la littérature sont insuffisantes pour établir une corrélation entre les taux d’anticorps et la gravité de la pathologie.
L’incidence varie de 2,4 à 21,7 cas par million de personnes, touchant principalement les femmes âgées de plus de 60 ans.11 Les facteurs déclenchants de la pemphigoïde bulleuse englobent les traumatismes locaux et les causes iatrogènes médicamenteuses (furosémide, antimicrobiens, anti-inflammatoires non stéroïdiens, inhibiteurs des points de contrôle immunitaires, etc.). La pemphigoïde bulleuse est également associée à certaines affections neurologiques telles que la sclérose en plaques et la maladie de Parkinson.
Le pronostic de la pemphigoïde bulleuse reste relativement bénin comparé au pemphigus, malgré l’impact significatif sur la qualité de vie durant la phase active de la pathologie, notamment en raison des effets indésirables de la corticothérapie systémique.
Chez les patients atteints d’une forme de pemphigoïde bulleuse orale localisée, le traitement de première intention est la corticothérapie locale, facile d’utilisation et limitant les effets indésirables. L’optimisation de l’hygiène buccale et des soins parodontaux favorise une wmeilleure réponse au traitement, grâce au contrôle de l’inflammation gingivale induite par la plaque. En cas d’échec, les options thérapeutiques envisagées en seconde intention comprennent le tacrolimus local, le mycophénolate mofétil ou la tétracycline associée au nicotinamide.
La corticothérapie systémique, traitement principal depuis les années 1950, et les agents immunosuppresseurs sont indiqués dans les formes plus étendues. La disparition des bulles est généralement obtenue dans les quatre semaines après le début du traitement, permettant la diminution progressive de la posologie chez 60 à 90 % des patients. Cependant, la chronicité de la maladie peut nécessiter une corticothérapie au long cours, entraînant des effets indésirables.
Les immunosuppresseurs tels que l’azathioprine, le chlorambucil, la dapsone et le méthotrexate ont démontré leur efficacité dans le traitement de la pemphigoïde bulleuse, offrant une alternative à la corticothérapie de longue durée. Néanmoins, tous ces médicaments peuvent causer des effets indésirables graves, et les preuves de leur efficacité restent limitées.2,10
Dans les formes de pemphigoïde bulleuse réfractaire, de nouvelles approches thérapeutiques semblent prometteuses : le rituximab, un anticorps anti-CD20, a montré des résultats encourageants, avec des taux de rémission de 60 à 70 %.2,13-15 Les traitements par antirécepteur néonatal Fc (FcRn) ont également obtenu des résultats prometteurs chez les souris et semblent réguler les taux d’IgG de l’hôte, y compris les IgG pathogènes impliquées dans les maladies bulleuses auto-immunes.2 L’ixékizumab, un anticorps humanisé ciblant l’IL-17, est en cours d’évaluation.2 L’omalizumab a donné des résultats intéressants, confirmant le rôle pathogène des IgE dans le développement de la pemphigoïde bulleuse.7 En outre, des anticorps monoclonaux ciblant l’IL-5 font l’objet d’investigations et le diméthylfumarate est étudié pour ses effets anti-inflammatoires pléiotropiques.7
Le diagnostic repose sur l’examen histologique d’un échantillon de bulle frais, objectivant un clivage sous-épidermique associé à un infiltrat inflammatoire.5,7 L’immunofluorescence directe est la technique de référence, montrant un dépôt linéaire d’immunoglobulines G (IgG), A (IgA) ou de fractions du complément (C3) le long de la membrane basale épithéliale.1,2,4,8-10 Des tests immunosérologiques (test ELISA mesurant les taux sériques d’anticorps dirigés contre les protéines BP180 et BP230) sont couramment employés comme outils diagnostiques.1,2,8,10 Cependant, les données de la littérature sont insuffisantes pour établir une corrélation entre les taux d’anticorps et la gravité de la pathologie.
L’incidence varie de 2,4 à 21,7 cas par million de personnes, touchant principalement les femmes âgées de plus de 60 ans.11 Les facteurs déclenchants de la pemphigoïde bulleuse englobent les traumatismes locaux et les causes iatrogènes médicamenteuses (furosémide, antimicrobiens, anti-inflammatoires non stéroïdiens, inhibiteurs des points de contrôle immunitaires, etc.). La pemphigoïde bulleuse est également associée à certaines affections neurologiques telles que la sclérose en plaques et la maladie de Parkinson.
Le pronostic de la pemphigoïde bulleuse reste relativement bénin comparé au pemphigus, malgré l’impact significatif sur la qualité de vie durant la phase active de la pathologie, notamment en raison des effets indésirables de la corticothérapie systémique.
Chez les patients atteints d’une forme de pemphigoïde bulleuse orale localisée, le traitement de première intention est la corticothérapie locale, facile d’utilisation et limitant les effets indésirables. L’optimisation de l’hygiène buccale et des soins parodontaux favorise une wmeilleure réponse au traitement, grâce au contrôle de l’inflammation gingivale induite par la plaque. En cas d’échec, les options thérapeutiques envisagées en seconde intention comprennent le tacrolimus local, le mycophénolate mofétil ou la tétracycline associée au nicotinamide.
La corticothérapie systémique, traitement principal depuis les années 1950, et les agents immunosuppresseurs sont indiqués dans les formes plus étendues. La disparition des bulles est généralement obtenue dans les quatre semaines après le début du traitement, permettant la diminution progressive de la posologie chez 60 à 90 % des patients. Cependant, la chronicité de la maladie peut nécessiter une corticothérapie au long cours, entraînant des effets indésirables.
Les immunosuppresseurs tels que l’azathioprine, le chlorambucil, la dapsone et le méthotrexate ont démontré leur efficacité dans le traitement de la pemphigoïde bulleuse, offrant une alternative à la corticothérapie de longue durée. Néanmoins, tous ces médicaments peuvent causer des effets indésirables graves, et les preuves de leur efficacité restent limitées.2,10
Dans les formes de pemphigoïde bulleuse réfractaire, de nouvelles approches thérapeutiques semblent prometteuses : le rituximab, un anticorps anti-CD20, a montré des résultats encourageants, avec des taux de rémission de 60 à 70 %.2,13-15 Les traitements par antirécepteur néonatal Fc (FcRn) ont également obtenu des résultats prometteurs chez les souris et semblent réguler les taux d’IgG de l’hôte, y compris les IgG pathogènes impliquées dans les maladies bulleuses auto-immunes.2 L’ixékizumab, un anticorps humanisé ciblant l’IL-17, est en cours d’évaluation.2 L’omalizumab a donné des résultats intéressants, confirmant le rôle pathogène des IgE dans le développement de la pemphigoïde bulleuse.7 En outre, des anticorps monoclonaux ciblant l’IL-5 font l’objet d’investigations et le diméthylfumarate est étudié pour ses effets anti-inflammatoires pléiotropiques.7
Références
1. Rashid H, Lamberts A, Diercks GFH, Pas HH, Meijer JM, Bolling MC, et al. Oral lesions in autoimmune bullous diseases: An overview of clinical characteristics and diagnostic algorithm. Am J Clin Dermatol 2019;20(6):847‑61.
2. Hussain MH, Tanweer F, Sakagiannis G, Mair M, Mahmood S, Ashokkumar S. Pemphigus vulgaris and bullous pemphigoid of the upper aerodigestive tract: A review article and novel approaches to management. ORL 2021;83(6):395‑403.
3. Parlatescu I, Tovaru S, Tofan C, Perlea P, Milanesi E, Dobre M, et al. Gingival manifestations in oral chronic autoimmune bullous diseases: A retrospective study. Medicina 2024;60(1):167.
4. Tyldesley WR. Oral pemphigoid. Br J Oral Maxillofac Surg 1985;23(3):155‑66.
5. Shklar G, McCarthy PL. Oral lesions of mucous membrane pemphigoid: A study of 85 cases. Arch Otolaryngol 1971;93(4):354‑64.
6. Bilgic A, Aydin F, Sumer P, Keskiner I, Koc S, Bozkurt S, et al. Oral health related quality of life and disease severity in autoimmune bullous disease. Niger J Clin Pract 2020;23(2):159-64.
7. Giannetti L, Murri Dello Diago A. Therapy of autoimmune mouth bullous disease: 2020 review. Dermatol Ther 2021;34(1):e14376.
8. Carey B, Setterfield J. Mucous membrane pemphigoid and oral blistering diseases. Clin Exp Dermatol 2019;44(7):732‑9.
9. Carey B, Joshi S, Abdelghani A, Mee J, Andiappan M, Setterfield J. The optimal oral biopsy site for diagnosis of mucous membrane pemphigoid and pemphigus vulgaris. Br J Dermatol 2020;182(3):747‑53.
10. Alramadhan SA, Islam MN. Vesiculobullous lesions of the oral cavity. Oral Maxillofac Surg Clin North Am 2023;35(2):203‑17.
11. Al Ismaili A, Al Busaidi K, Nalawade T, Saraf S. Immune-mediated skin disorders and their oral manifestations in the Omani population: A hospital-based study. Oman Med J 2020;35(1):e84.
12. Fässler M, Rammlmair A, Feldmeyer L, Suter VGA, Gloor AD, Horn M, et al. Mucous membrane pemphigoid and lichenoid reactions after immune checkpoint inhibitors: Common pathomechanisms. J Eur Acad Dermatol Venereol 2020;34(2):e112-5.
13. Persson MS, Harman KE, Thomas KS, Chalmers JR, Vinogradova Y, Langan SM, et al. Long-term oral prednisolone exposure in primary care for bullous pemphigoid: Population-based study. Br J Gen Pract 2021;71(713):e904‑11.
14. Leuci S, Ruoppo E, Adamo D, Calabria E, Mignogna MD. Oral autoimmune vesicobullous diseases: Classification, clinical presentations, molecular mechanisms, diagnostic algorithms, and management. Periodontol 2000 2019;80(1):77‑88.
15. Buonavoglia A, Leone P, Dammacco R, Di Lernia G, Petruzzi M, Bonamonte D, et al. Pemphigus and mucous membrane pemphigoid: An update from diagnosis to therapy. Autoimmun Rev 2019;18(4):349‑58.
2. Hussain MH, Tanweer F, Sakagiannis G, Mair M, Mahmood S, Ashokkumar S. Pemphigus vulgaris and bullous pemphigoid of the upper aerodigestive tract: A review article and novel approaches to management. ORL 2021;83(6):395‑403.
3. Parlatescu I, Tovaru S, Tofan C, Perlea P, Milanesi E, Dobre M, et al. Gingival manifestations in oral chronic autoimmune bullous diseases: A retrospective study. Medicina 2024;60(1):167.
4. Tyldesley WR. Oral pemphigoid. Br J Oral Maxillofac Surg 1985;23(3):155‑66.
5. Shklar G, McCarthy PL. Oral lesions of mucous membrane pemphigoid: A study of 85 cases. Arch Otolaryngol 1971;93(4):354‑64.
6. Bilgic A, Aydin F, Sumer P, Keskiner I, Koc S, Bozkurt S, et al. Oral health related quality of life and disease severity in autoimmune bullous disease. Niger J Clin Pract 2020;23(2):159-64.
7. Giannetti L, Murri Dello Diago A. Therapy of autoimmune mouth bullous disease: 2020 review. Dermatol Ther 2021;34(1):e14376.
8. Carey B, Setterfield J. Mucous membrane pemphigoid and oral blistering diseases. Clin Exp Dermatol 2019;44(7):732‑9.
9. Carey B, Joshi S, Abdelghani A, Mee J, Andiappan M, Setterfield J. The optimal oral biopsy site for diagnosis of mucous membrane pemphigoid and pemphigus vulgaris. Br J Dermatol 2020;182(3):747‑53.
10. Alramadhan SA, Islam MN. Vesiculobullous lesions of the oral cavity. Oral Maxillofac Surg Clin North Am 2023;35(2):203‑17.
11. Al Ismaili A, Al Busaidi K, Nalawade T, Saraf S. Immune-mediated skin disorders and their oral manifestations in the Omani population: A hospital-based study. Oman Med J 2020;35(1):e84.
12. Fässler M, Rammlmair A, Feldmeyer L, Suter VGA, Gloor AD, Horn M, et al. Mucous membrane pemphigoid and lichenoid reactions after immune checkpoint inhibitors: Common pathomechanisms. J Eur Acad Dermatol Venereol 2020;34(2):e112-5.
13. Persson MS, Harman KE, Thomas KS, Chalmers JR, Vinogradova Y, Langan SM, et al. Long-term oral prednisolone exposure in primary care for bullous pemphigoid: Population-based study. Br J Gen Pract 2021;71(713):e904‑11.
14. Leuci S, Ruoppo E, Adamo D, Calabria E, Mignogna MD. Oral autoimmune vesicobullous diseases: Classification, clinical presentations, molecular mechanisms, diagnostic algorithms, and management. Periodontol 2000 2019;80(1):77‑88.
15. Buonavoglia A, Leone P, Dammacco R, Di Lernia G, Petruzzi M, Bonamonte D, et al. Pemphigus and mucous membrane pemphigoid: An update from diagnosis to therapy. Autoimmun Rev 2019;18(4):349‑58.
Une question, un commentaire ?
Sur le même thème
Exercice
Exercice
Article