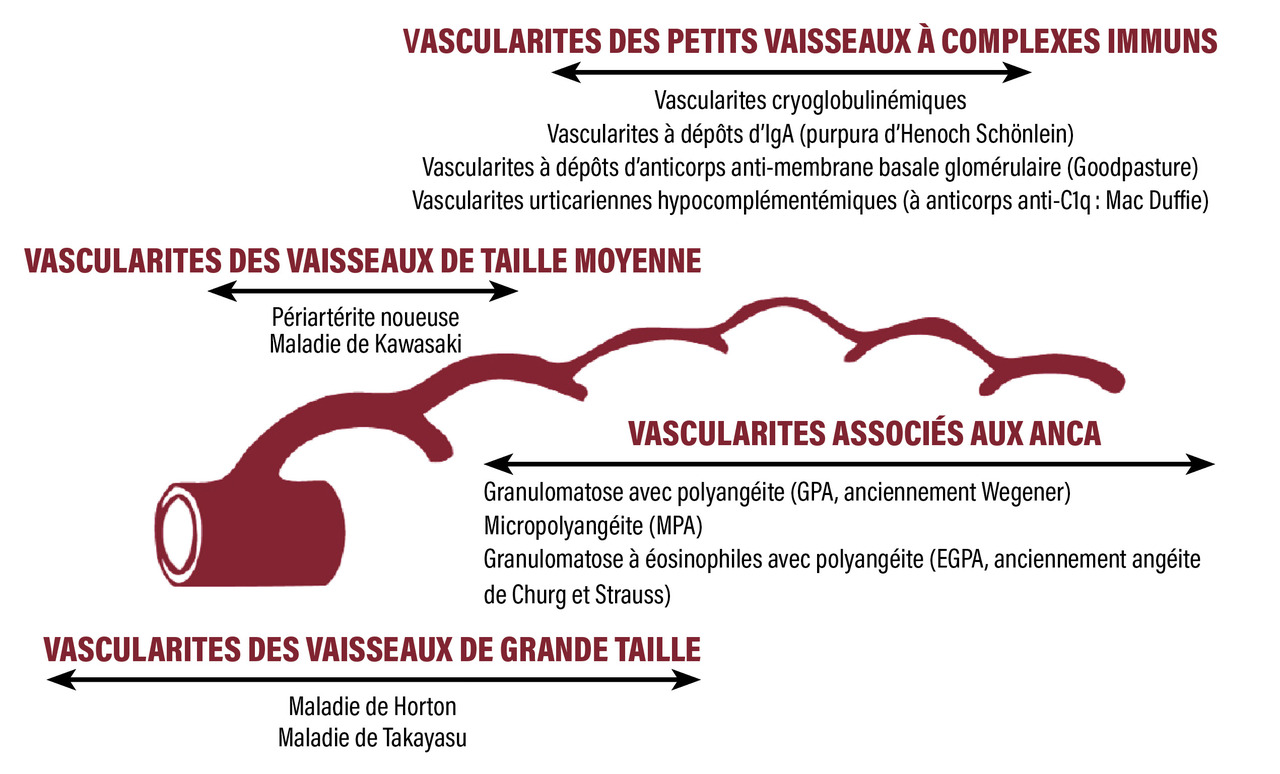
Les périartérites noueuses (PAN) sont des maladies systémiques appartenant au groupe des vascularites nécrosantes systémiques (VNS). Conformément à la classification des vascularites de Chapel Hill (fig. 1),1 la PAN est considérée comme une vascularite des moyens vaisseaux, au même titre que la maladie de Kawasaki. Cela signifie qu’elle atteint principalement les artères de moyen calibre, à savoir les principales artères viscérales et leurs branches. Ainsi, elle ne concerne pas les artérioles, capillaires ou veinules, et ne donne pas de glomérulonéphrite.1
Probables dépôts de complexes immuns et auto-inflammation
Les PAN constituent vraisemblablement un groupe de pathologies dont les mécanismes sont divers en fonction de leur cause, primitive ou secondaire.2 Elles sont probablement associées à des dépôts de complexes immuns dans la paroi des artères de moyen calibre, avec activation du complément et recrutement des cellules de l’inflammation. Ce mécanisme est clairement impliqué dans les PAN associées au virus de l’hépatite B (VHB), mais demeure incertain dans les autres formes de PAN.3
On observe histologiquement un infiltrat focal polymorphe (polynucléaires neutrophiles [PNN], polynucléaires éosinophiles [PNE], macrophages, lymphocytes T) dans la paroi des artères de moyen calibre, préférentiellement au niveau de la média, sans visualiser de granulome. Ces infiltrats conduisent à une nécrose transmurale, avec cicatrice fibrineuse qui entraîne sténoses et microanévrismes. Il est fréquent d’observer des lésions d’âge différent lors des biopsies.3,4
En dehors des dépôts de complexes immuns, un mécanisme auto-inflammatoire associé est fortement suspecté. En effet, les profils cytokiniques des patients retrouvent volontiers une élévation du tumor necrosis factor (TNF) α, et des interleukines 6 (IL- 6) et 1β. De plus, les descriptions récentes de PAN secondaires à des syndromes auto-inflammatoires tels que la fièvre méditerranéenne familiale, le déficit en adénosine désaminase 2 (DADA2) et le VEXAS tendent à plaider en faveur de ce mécanisme associé.3
Maladie rare, primitive ou secondaire
Au même titre que les autres vascularites nécrosantes systémiques (VNS), il s’agit d’une maladie rare, avec une incidence estimée entre 7 et 63 cas/an/million d’habitants.4 Si l’âge médian des patients au diagnostic est de 51 ans, elle peut débuter à tout âge.5,6 Il existe une prépondérance masculine (H/F = 2/1).
On distingue les PAN cutanées, avec une vascularite ne concernant que la peau,7 des PAN systémiques, dont les manifestations sont plus globales. Les PAN systémiques sont classées en deux grands groupes étiologiques : primitives, quand aucune cause associée n’est retrouvée ; ou secondaires. Parmi les PAN secondaires, celles associées au virus de l’hépatite B (VHB) constituaient un tiers des PAN rapportées dans une série française de 348 PAN colligées de 1963 à 2005.5 Cette proportion s’est effondrée, puisqu’une nouvelle série française de 196 PAN colligées depuis 2005 ne rapportait aucune PAN associée au VHB.6 Parmi les 28 % de PAN secondaires, celles-ci étaient associées à un syndrome myélodysplasique ou une leucémie myélomonocytaire chronique, un cancer solide, une hémopathie lymphoïde, une maladie auto-inflammatoire (telle que la fièvre méditerranéenne familiale), un syndrome hyperéosinophilique, une infection par le virus de l’immunodéficience humaine (VIH) ou par le virus de l’hépatite C (VHC).
Récemment ont été décrites deux maladies dont les manifestations peuvent ressembler à celles d’une PAN et qui doivent être différenciées des PAN secondaires :
le DADA2,8,9 maladie génétique à transmission autosomique récessive, touchant principalement le sujet jeune, avec un diagnostic porté en moyenne à l’âge de 15 ans. Il se manifeste par un tableau fébrile, une vascularite des moyens vaisseaux ressemblant à une PAN avec atteintes cutanée et du système nerveux central (accidents vasculaires cérébraux ischémiques ou plus rarement hémorragiques). D’autres atteintes viscérales sont décrites, au même titre que dans la PAN. S’y associent fréquemment une lymphoprolifération avec adénopathies et hépatosplénomégalie, un déficit immunitaire et des cytopénies ;
le VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic)10,11 qui est un syndrome auto-inflammatoire acquis, en lien avec une mutation d’UBA1 (voir La Revue du Praticien, février 2025, pages 196 à 199). Il se manifeste aussi par un tableau fébrile associé à des signes cutanés, des chondrites, des thromboses et parfois par une vascularite des moyens vaisseaux évocatrice de PAN. Un syndrome myélodysplasique lui est fréquemment associé.
Manifestations cliniques variées
À l’instar des autres vascularites nécrosantes systémiques, les PAN se manifestent fréquemment par une franche altération de l’état général, notamment une perte de poids et de la fièvre, présentes chez deux tiers des patients (tableau).5,6
Fréquentes atteintes neurologiques
Les atteintes neurologiques sont les plus fréquentes de la PAN. Il s’agit le plus souvent de neuropathies périphériques, de survenue précoce dans la maladie, de type mononévrite multiple (fig. 2).12 Celle-ci est décrite chez deux patients sur trois ; elle est bien plus fréquente que la polyneuropathie. Elle se manifeste par des douleurs intenses, dans un ou plusieurs territoires tronculaires (branches du sciatique : fibulaires ou tibiales sont les plus souvent touchées).5 S’y associent rapidement des troubles sensitifs, puis, plus tardivement, une atteinte motrice. Ces atteintes tronculaires peuvent être multifocales, voire bilatérales, mais le plus souvent asynchrones.
Rares atteintes du système nerveux central
Les atteintes du système nerveux central (SNC) ne concernent que 5 à 9 % des patients mais grèvent le pronostic et constituent un critère de gravité. Il s’agit de lésions vasculaires cérébrales, principalement d’origine ischémique, attribuables soit à la vascularite, soit à l’hypertension artérielle et à l’hypercoagulabilité de patients inflammatoires. Les formes hémorragiques sont plus rares, en lien avec une transformation hémorragique d’un accident ischémique ou avec la rupture d’un microanévrisme. Il est fréquent que les patients décrivent des céphalées, voire une encéphalopathie ou une comitialité, pouvant parfois s’inscrire dans le cadre d’une leucoencéphalopathie postérieure réversible (posterior-reversible encephalopathy syndrome [PRES]).12 Ces atteintes du SNC doivent faire évoquer un DADA2, notamment en cas de forme juvénile ou s’il existe des infarctus lacunaires multiples du tronc cérébral, du cervelet ou des noyaux gris centraux.9
Arthralgies inflammatoires pour plus de la moitié des patients
Les PAN se manifestent dans plus de 50 % des cas par des arthralgies inflammatoires ainsi que par des myalgies, parfois intenses, et le plus souvent avec des CPK normales.5,6,13
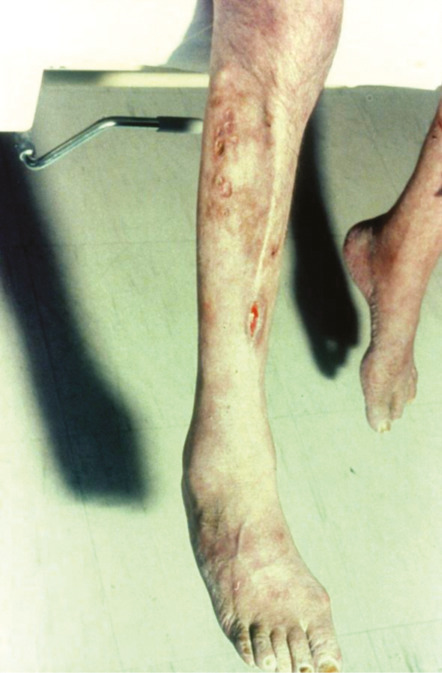
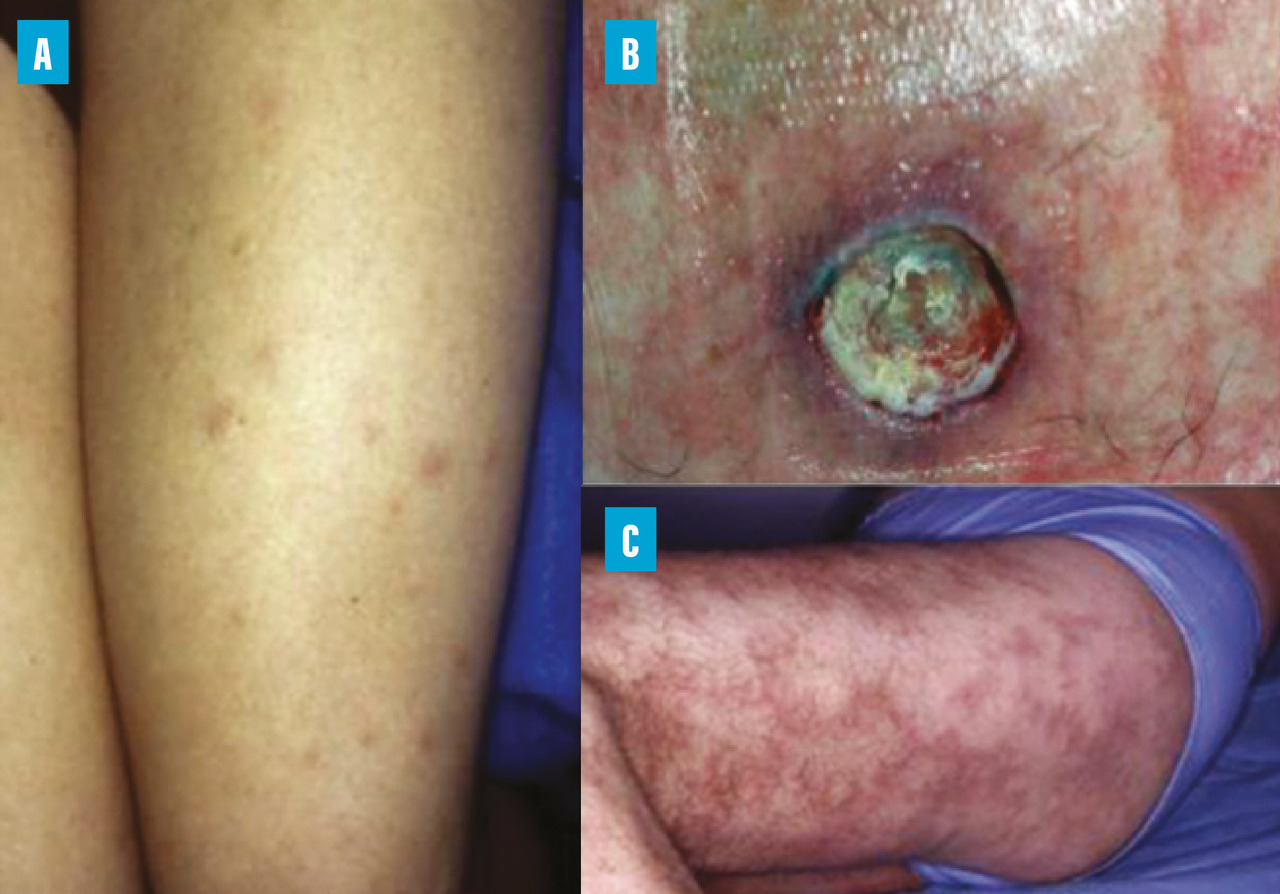
Manifestations cutanées parfois exclusives
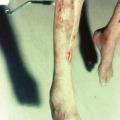
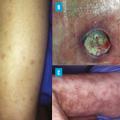
Les manifestations cutanées sont présentes chez la moitié des patients atteints de PAN systémiques. Purpura vasculaire, livedo, nodules et ulcérations sont les lésions les plus fréquentes (fig. 3). Il existe des PAN cutanées exclusives qui ne sont pas associées à une forme systémique de la maladie, excepté une atteinte neurologique périphérique fréquemment en regard des lésions cutanées.7 Dans ce contexte, en l’absence de neuropathie périphérique, la colchicine peut être une bonne option thérapeutique. En cas d’inefficacité, l’association de prednisone et d’azathioprine semble être la plus efficace.14
Atteintes digestives, rénales et cardiaques
Les atteintes digestives, rénales et cardiaques constituent des facteurs pronostiques de ces vascularites.
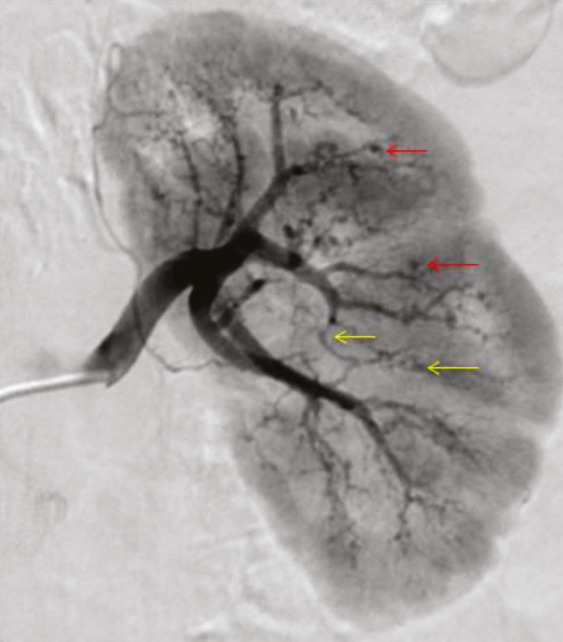
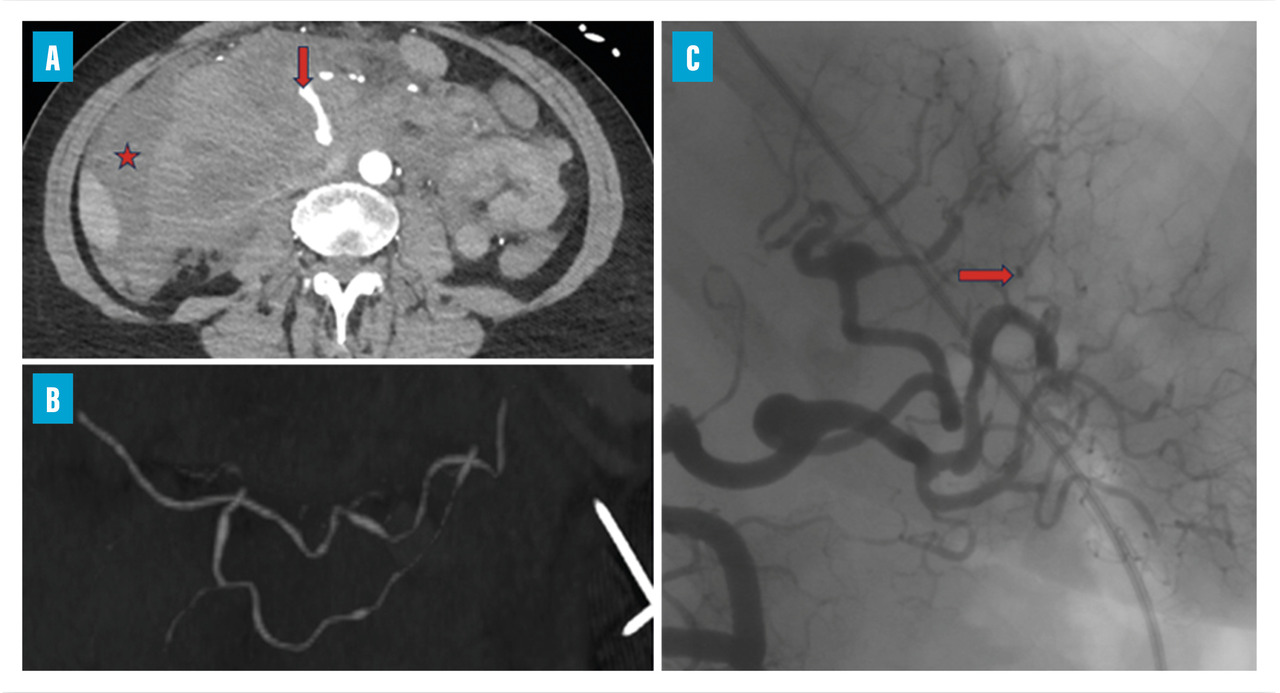
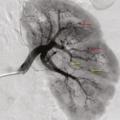
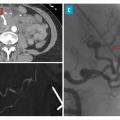
Les atteintes digestives et rénales sont vasculaires, en lien avec des sténoses des moyens vaisseaux, responsables de phénomènes ischémiques et/ou de microanévrismes (fig. 4).6 Leur diagnostic se confirme à l’imagerie, notamment par une angiotomodensitométrie (TDM). L’artériographie, examen de référence, peut s’avérer nécessaire, à visée diagnostique en cas de négativité de la TDM,2 mais surtout à visée thérapeutique, permettant une artério-embolisation en cas d’hémorragie grave.
Signes des atteintes digestives
Les douleurs abdominales, volontiers post-prandiales, sont fréquentes. Elles peuvent être révélatrices de complications sévères telles que les perforations digestives ou les hémorragies intrapéritonéales (fig. 5), gastro-intestinales ou sous-capsulaires. Ces complications s’observent chez 4 % des patients, de même que les cholécystites et pancréatites alithiasiques. Au total, jusqu’à 14 % des patients atteints de PAN peuvent subir un geste chirurgical pour une complication digestive.5,15
Signes des atteintes rénales
L’atteinte rénale se manifeste soit par une hypertension artérielle isolée, parfois sévère, soit par une authentique néphropathie vasculaire (insuffisance rénale aiguë, protéinurie tubulaire +/- hématurie microscopique, stigmates biologiques de microangiopathie thrombotique).16 Elle est en lien avec des sténoses, pouvant aussi entraîner des infarctus rénaux. Les microanévrismes des artères rénales et de leurs branches peuvent être responsables d’hématomes sous-capsulaires ou rétropéritonéaux.17 Si l’insuffisance rénale ne se voit que dans 12 % des PAN,5,6 cette néphropathie vasculaire, non glomérulaire, représente un enjeu diagnostique majeur. Contrairement aux autres vascularites, notamment celles associées aux anticorps anticytoplasme des polynucléaires neutrophiles (antineutrophil cytoplasmic antibodies [ANCA]), la biopsie rénale ne doit pas être réalisée avant d’avoir exclu les microanévrismes en artériographie, au risque de complications hémorragiques.16,18
Deux entités d’atteinte cardiaque
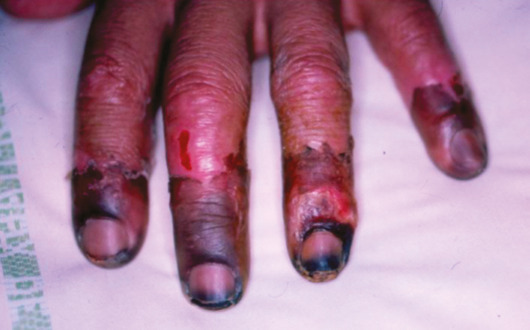
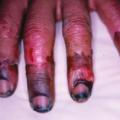
Concernant l’atteinte cardiaque, il existe principalement deux entités : l’artérite coronarienne, avec sténose et/ou anévrismes,19 responsable de syndromes coronariens ; les myocardites, pour lesquelles l’imagerie par résonance magnétique (IRM) cardiaque permet le diagnostic, la biopsie endomyocardique pouvant s’avérer utile en cas de doute.20 Une péricardite isolée ou associée peut être observée dans 5 % des PAN.5Sur le plan vasculaire, des ischémies, voire des nécroses des extrémités, sont présentes chez 12 % des patients (fig. 6). Le Doppler artériel peut révéler des occlusions des artères distales telles que les tibiales et fibulaires. L’angio-TDM, ou l’artériographie si nécessaire, confirme dans ce contexte des sténoses irrégulières, des anévrismes et/ou des occlusions multiples.21
Autres manifestations
L’orchite est une manifestation fortement évocatrice de PAN. Elle peut être asymptomatique ou responsable de douleurs testiculaires. Elle peut rarement se compliquer d’une nécrose testiculaire.22Les atteintes ophtalmiques sont des rétinopathies hypertensives, des vascularites rétiniennes mais aussi, parfois, des kératites ulcérantes périphériques, des sclérites nécrosantes, des épisclérites et des uvéites.5,23L’atteinte pulmonaire est non spécifique, avec des infiltrats ou des sérites, potentiellement associées à l’atteinte myocardique, à l’hypertension artérielle sévère ou à l’hyperéosinophilie.5,6Biologiquement, il existe un syndrome inflammatoire biologique majeur avec une protéine C réactive (CRP) s’élevant à 120 mg/L en moyenne, une hyperleucocytose neutrophilique et une monocytose fréquentes. Il existe une hyperéosinophilie chez près d’un malade sur dix.6 La recherche d’ANCA est négative.
Diagnostic
Le diagnostic de PAN se fait par l’histologie d’un organe atteint et/ou par l’imagerie vasculaire.
Biopsie selon la localisation
La réalisation d’une biopsie ne doit pas retarder le traitement en cas de forte suspicion diagnostique et elle doit prendre en compte le rapport bénéfice/risque.4
La biopsie rénale est contre-indiquée en cas de suspicion de PAN. En effet, l’atteinte rénale de la PAN est de type vasculaire, et non glomérulaire ni interstitielle, ce qui rend la rentabilité diagnostique faible. De plus, les possibles microanévrismes rénaux et l’hypertension artérielle, fréquente dans ce contexte, augmentent drastiquement le risque hémorragique d’une biopsie rénale.4
Quand elle peut être réalisée, l’histologie révèle une atteinte des artères de moyen calibre, avec infiltrat de cellules inflammatoires polymorphes, et une nécrose fibrinoïde transmurale, sans granulome ni cellule géante.2
La peau est un site privilégié, mais la biopsie doit être profonde et contenir de l’hypoderme (technique en « fuseau » ou en « double punch »).
La biopsie musculaire peut être utile. La biopsie neuromusculaire guidée par l’électroneuromyogramme est plus rentable pour établir le diagnostic mais peut laisser des séquelles à type de dysesthésies dans le territoire biopsié.
Imagerie artérielle non invasive en première intention
L’imagerie artérielle peut suffire à poser le diagnostic, en cas de suspicion clinique et de présence d’anévrismes sacciformes ou fusiformes, de sténoses des artères de moyen calibre, notamment rénales, hépatiques ou digestives.2,4 En première intention, une imagerie non invasive telle que l’angio-TDM ou l’angio-IRM (en cas de contre-indication aux produits de contraste iodés) est recommandée mais nécessite un regard exercé de la part du radiologue. L’artériographie reste l’examen de référence en cas de doute diagnostique, mais n’est réalisée qu’en deuxième intention, compte tenu d’éventuelles complications, à type d’hématome, de faux anévrismes au point de ponction ou de néphropathie aux produits de contraste.2 Elle peut aussi jouer un rôle thérapeutique, en cas de saignement sévère, permettant de réaliser une embolisation.
Éliminer les diagnostics différentiels
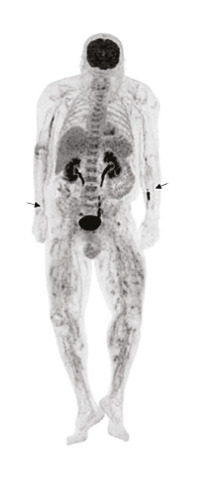
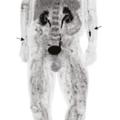
L’élimination des diagnostics différentiels est importante pour retenir le diagnostic de PAN. La fréquente altération de l’état général présente chez les patients doit conduire en premier lieu à rechercher une néoplasie solide, une hémopathie maligne ou une infection de type tuberculose, endocardite, etc.4 La réalisation d’une tomodensitométrie thoraco-abdomino-pelvienne (TDM-TAP) avec temps artériel, permet de rechercher ces diagnostics différentiels mais peut aussi révéler les sténoses, occlusions et anévrismes des moyens vaisseaux. La tomographie par émission de positons au fluorodésoxyglucose (TEP-FDG) peut avoir le même rôle de recherche de ces diagnostics différentiels et peut révéler une hyperfixation des artères de moyen calibre, associée à une hyperfixation musculaire, fortement évocatrice de PAN (fig. 7).24 Les autres vascularites constituent aussi des diagnostics alternatifs. Notamment la présence d’ANCA ou d’une cryoglobulinémie à taux significatif a tendance à exclure le diagnostic de PAN.1,25
D’autres examens systématiques permettent d’éliminer les diagnostics différentiels ou de retenir le diagnostic de PAN secondaire :4
- sérologies virales (VIH, VHB, VHC) ;
- recherche d’infection bactérienne par hémocultures, échocardiographie cardiaque, sérologies Coxiella, Bartonella, syphilis ;
- recherche d’arguments cliniques et iconographiques (TDM) pour une tuberculose, une néoplasie, une hémopathie ;
- bilan immunologique pour éliminer une autre maladie auto-immune avec dosages des ANCA, de la cryoglobulinémie, des facteurs antinucléaires, de C3 et C4, du facteur rhumatoïde, des anti-B2GP1, anticardiolipine et anticoagulant circulant ;
- recherche d’argument pour une maladie de Behçet, notamment une aphtose bipolaire, des thromboses veineuses de sites atypiques, des uvéites ;
- recherche d’argument pour un déficit en ADA2 ;
- recherche d’argument pour une myélodysplasie ou un VEXAS (séquençage à haut débit ou next-generation sequencing [NGS] myéloïde).
En cas de suspicion de PAN sans preuve histologique, la présence d’un des trois critères (hépatite B avec présence de l’antigène ou de l’ADN sérique, anomalies artériographiques, mononeuropathie) et l’absence de cinq critères (ANCA, cryoglobuline, asthme, signes ORL, glomérulonéphrite) permettraient de retenir le diagnostic de PAN avec une spécificité de 92 %.25
Maladie grave de pronostic sombre, variable selon la cause
Avec une mortalité estimée à 25 % à cinq ans,5 les PAN constituent un groupe de maladies particulièrement graves.
Cette sévérité dépend de la cause de la PAN : liée au VHB (mortalité à cinq ans de 34 % versus 20 % pour les non liées),5 secondaire (mortalité à 5 ans trois fois plus élevée).6 Les autres facteurs pronostiques sont cliniques, représentés notamment par le five factor score (FFS) établi en 1996.26 Chaque item de ce score représente un critère négativement associé à la survie des patients : protéinurie > 1 g/24 h, créatininémie > 140 µM/L, cardiomyopathie spécifique symptomatique, atteinte digestive grave (perforation, hémorragie, pancréatite), atteinte du système nerveux central.
Neuf PAN sur dix répondent à une première ligne de traitement. L’association à une myélodysplasie, l’atteinte digestive, cutanée ou rénale, constituent des risques d’échec thérapeutique en première ligne.6
Les rechutes sont fréquentes : 55 % des patients font au moins une rechute à cinq ans.6 Les atteintes digestives et cutanées sont associées à des rechutes plus fréquentes.
Traitement selon les causes et les critères de gravité
Le traitement des PAN systémiques se fonde sur les recommandations françaises (Protocole national de diagnostic et de soins [PNDS] 2019).4 Il doit prendre en compte :
- la cause, notamment secondaire, liée au VHB ou non ;
- les critères de gravité, fondés sur le FFS 1996.26
PAN primitives sans facteur de mauvais pronostic
Les PAN primitives, sans facteur de mauvais pronostic (FFS = 0) peuvent se traiter par corticoïdes seuls, sous réserve d’une bonne tolérance. La prednisone, à la dose de 1 mg/kg, est le traitement de référence. Une décroissance est réalisée à partir de trois semaines de traitement pour obtenir un sevrage dans les six mois à deux ans. L’adjonction d’un immunosuppresseur, tel que l’azathioprine, n’a pas montré de supériorité en première intention dans ce contexte.27 Un immunosuppresseur est néanmoins introduit en cas de corticodépendance (plus de 7,5 à 10 mg/j), en privilégiant l’azathioprine ou le méthotrexate.
PAN primitives avec facteurs de mauvais pronostic
Les PAN primitives, associées à un ou plusieurs facteurs de mauvais pronostic (FFS > 0), nécessitent l’adjonction d’un immunosuppresseur à la corticothérapie. Dans ce contexte, des bolus intraveineux de méthylprednisolone (7,5 à 15 mg/kg à J1, J2 et J3) sur trois jours sont privilégiés.2 Le cyclophosphamide intraveineux est l’immunosuppresseur de référence. Il s’injecte à J1, J15 et J29 à la dose de 0,6 g/m2, puis toutes les trois semaines pour un total de 6 à 9 injections, à la dose de 0,7 g/m2, sans dépasser 1 200 mg. Une dose de 500 mg par injection chez la personne de plus de 65 ans permet de réduire le risque d’effets indésirables, sans perte d’efficacité.28 Après obtention d’une rémission, entre deux et quatre semaines après la dernière injection de cyclophosphamide, un relais est réalisé par azathioprine per os (2 à 3 mg/kg/j) sans dépasser 200 mg/j ou par méthotrexate per os ou sous-cutané (0,3 mg/kg/semaine) pendant douze à dix-huit mois.
PAN réfractaires et secondaires
Dans les cas de PAN réfractaires, il existe peu de thérapeutiques ayant montré leur efficacité, mais certains cas de succès thérapeutique sont rapportés sous tocilizumab, anti-TNF alpha ou rituximab.29
Les PAN associées au VHB requièrent l’association de prednisone à une dose comprise entre 0,5 et 1 mg/kg/j, d’antiviraux (ténofovir ou entécavir) et d’échanges plasmatiques.
Pour les autres PAN secondaires, la prise en charge est moins codifiée, mais le traitement de la pathologie associée (myélodysplasie, cancer, etc.) peut permettre d’obtenir une rémission.6 Les déficits en ADA2 sont préférentiellement traités par anti-TNF alpha.2,8
Mesures pour limiter les effets indésirables
Des mesures associées sont indispensables pour limiter les effets indésirables des thérapeutiques :
- prophylaxie antipneumocystose par cotrimoxazole ;
- vaccinations ;
- prise en charge des facteurs de risque vasculaire ;
- dépistage de cancers ;
- prévention de l’ostéoporose ;
- alimentation équilibrée (sans régime strict) ;
- poursuite de l’exercice physique ;
- préservation de la fertilité (sous cyclophosphamide).
Pas de biomarqueur spécifique
Les périartérites noueuses sont des maladies rares et hétérogènes. Leur diagnostic est difficile et prend en compte les manifestations cliniques, les données histologiques ou une imagerie vasculaire. Il n’existe pas à ce jour de biomarqueur spécifique. Si les atteintes les plus fréquentes sont cutanées ou du système nerveux périphérique, le pronostic est conditionné par les atteintes rénales, digestives, cardiaques ou du système nerveux central, ainsi que par le caractère secondaire de la PAN. Les corticoïdes, le cyclophosphamide et l’azathioprine restent les thérapeutiques préférentielles. L ’utilisation des biothérapies tient une place marginale, faute d’une efficacité suffisante démontrée.
2. Chung SA, Gorelik M, Langford CA, et al. 2021 American College of Rheumatology/Vasculitis Foundation Guideline for the management of polyarteritis nodosa. Arthritis Care Res 2021;73(8):1061‑70.
3. Wolff L, Horisberger A, Moi L, et al. Polyarteritis nodosa: Old disease, new etiologies. Int J Mol Sci 2023;24(23):16668.
4. Terrier B, Darbon R, Durel CA, et al. French recommendations for the management of systemic necrotizing vasculitides (polyarteritis nodosa and ANCA-associated vasculitides). Orphanet J Rare Dis 2020;15(2):351.
5. Pagnoux C, Seror R, Henegar C, et al. Clinical features and outcomes in 348 patients with polyarteritis nodosa: A systematic retrospective study of patients diagnosed between 1963 and 2005 and entered into the French Vasculitis Study Group Database. Arthritis Rheum 2010;62(2):616‑26.
6. Rohmer J, Nguyen Y, Trefond L, et al. Clinical features and long-term outcomes of patients with systemic polyarteritis nodosa diagnosed since 2005: Data from 196 patients. J Autoimmun 2023;139:103093.
7. Chasset F, Frances C. Cutaneous manifestations of medium- and large-vessel vasculitis. Clin Rev Allergy Immunol 2017;53(3):452‑68.
8. Schnappauf O, Sampaio Moura N, Aksentijevich I, et al. Sequence-based screening of patients with idiopathic polyarteritis nodosa, granulomatosis with polyangiitis, and microscopic polyangiitis for deleterious genetic variants in ADA2. Arthritis Rheumatol Hoboken NJ 2021;73(3):512‑9.
9. Barron KS, Aksentijevich I, Deuitch NT, et al. The spectrum of the deficiency of adenosine deaminase 2: An observational analysis of a 60 patients cohort. Front Immunol 2021;12:811473.
10. Georgin-Lavialle S, Terrier B, Guedon AF, et al. Further characterization of clinical and laboratory features in Vexas syndrome: Large-scale analysis of a multicentre case series of 116 French patients. Br J Dermatol 2022;186(3):564‑74.
11. Beck DB, Ferrada MA, Sikora KA, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med 2020;383(27):2628‑38.
12. de Boysson H, Guillevin L. Polyarteritis nodosa neurologic manifestations. Neurol Clin 2019;37(2):345‑57.
13. Ganeshanandan LR, Brusch AM, Dyke JM,et al. Polyarteritis nodosa isolated to muscles - A case series with a review of the literature. Semin Arthritis Rheum 2020;50(3):503‑8.
14. Bettuzzi T, Jachiet M, Sbidian E, et al. Efficacy and safety of treatments in cutaneous polyarteritis nodosa: A French observational retrospective study. J Am Acad Dermatol 2022;86(5):1035‑41.
15. Ebert EC, Kierson M, Hagspiel KD. Gastrointestinal and hepatic manifestations of sarcoidosis. Am J Gastroenterol 2008;103(12):3184‑92; quiz 3193.
16. Perrin J, Carvelli J, Gondouin B, et al. Beware, polyarteritis nodosa still exists in nephrology! Nephrol Ther 2016;12(6):463‑7.
17. Agarwal A, Bansal M, Pandey R, et al. Bilateral subcapsular and perinephric hemorrhage as the initial presentation of polyarteritis nodosa. Intern Med Tokyo Jpn 2012;51(9):1073‑6.
18. Howard T, Ahmad K, Swanson JAA, et al. Polyarteritis nodosa. Tech Vasc Interv Radiol 2014;17(4):247‑51.
19. Schrader ML, Hochman JS, Bulkley BH. The heart in polyarteritis nodosa: A clinicopathologic study. Am Heart J 1985;109(6):1353‑9.
20. Chimenti C, Alfarano M, Toto F, et al. Myocarditis and intramural coronary vasculitis in polyarteritis nodosa: An unusual treatable form of heart failure. ESC Heart Fail 2020;7(6):4357‑60.
21. Shukla A, Aggarwal A. Polyarteritis nodosa presenting as peripheral vascular disease and acute limb ischemia. J Postgrad Med 2017;63(1):47‑9.
22. Toepfer NJ, Lountzis NI, Ugoeke JC, et al. Polyarteritis nodosa with bilateral asynchronous testicular necrosis: A case report. Case Rep Urol 2011;2011:465353.
23. Carreño E, Olivas-Vergara OM. Systemic vasculitis and its association with the eye. Ophthalmologica 2023;246(3‑4):174‑80.
24. Fagart A, Machet T, Collet G, Q et al. Fluorodeoxyglucose positron emission tomography-computed tomography findings in a first series of 10 patients with polyarteritis nodosa. Rheumatol Oxf Engl 2022;61(4):1663‑8.
25. Henegar C, Pagnoux C, Puéchal X, et al. A paradigm of diagnostic criteria for polyarteritis nodosa: Analysis of a series of 949 patients with vasculitides.Arthritis Rheum 2008;58(5):1528‑38.
26. Guillevin L, Lhote F, Gayraud M, et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore) 1996;75(1):17‑28.
27. Puechal X, Pagnoux C, Baron G, et al. Adding azathioprine to remission-induction glucocorticoids for eosinophilic granulomatosis with polyangiitis (Churg-Strauss), microscopic polyangiitis, or polyarteritis nodosa without poor prognosis factors: A randomized, controlled trial. Arthritis Rheumatol Hoboken NJ 2017;69(11):2175‑86.
28. Pagnoux C, Quemeneur T, Ninet J, et al. Treatment of systemic necrotizing vasculitides in patients aged sixty-five years or older: Results of a multicenter, open-label, randomized controlled trial of corticosteroid and cyclophosphamide-based induction therapy. Arthritis Rheumatol Hoboken NJ 2015;67(4):1117‑27.
29. Hadjadj J, Canzian A, Karadag O, et al. Use of biologics to treat relapsing and/or refractory polyarteritis nodosa: Data from a European collaborative study. Rheumatol Oxf Engl 2022;62(1):341‑6.