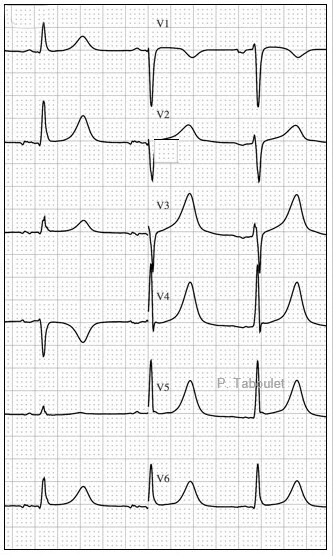
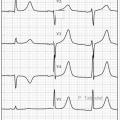
Le syndrome du QT long (SQTL) [figure] désigne une anomalie de la repolarisation ventriculaire d’origine génétique consistant en un allongement de l’intervalle QT sur l’électrocardiogramme (ECG) à 12 dérivations, exposant les patients qui en sont atteints à un risque significatif de syncope ou de mort subite dû à des troubles du rythme ventriculaire graves (torsades de pointes ou fibrillation ventriculaire). Il a une prévalence d’environ 1 naissance vivante sur 2 000 à 2 500.
La transmission de ce syndrome est généralement de type autosomique dominant (risque de transmission de 50 % à chaque naissance), impliquant la nécessité d’organiser le dépistage familial pour permettre une prise en charge précoce.
Signes évocateurs
Il faut y penser devant une syncope, ou en cas de mort subite d’un parent proche. Le diagnostic repose sur la mesure de l’intervalle QT corrigé (QTc, en utilisant la formule de Bazett) sur l’électrocardiogramme en 12 dérivations et des examens complémentaires. La vérification de l’ionogramme (avec dosage correct de la kaliémie : prélèvement à jeun, au repos, sans massage ni mouvement du bras du côté du prélèvement, et sans garrot) doit être systématique.
Un bilan génétique est indispensable pour analyser, au moins, les 3 principaux gènes impliqués (près de 90 % des cas sont associés à une anomalie génétique dans un de ces gènes). Il nécessite une consultation de cardiogénétique avec prélèvements réalisés dans l’un des centres de référence ou de compétences indiqués en annexe 2 du Protocole national de diagnostic et de soins. Les délais de réponse sont longs (plusieurs mois) et l’absence de mutation dans l’un des 3 gènes n’élimine pas le diagnostic de SQTL, compte tenu de l’existence de formes génétiques très rares.
Le score de Schwartz modifié peut aider à évaluer le risque de SQTL. Il a été actualisé dans les recos 2022 de la Société européenne de cardiologie (ESC) [tableau]. Un QTc ≥ 480 ms ou une mutation pathogénique reconnue (gènes KCNQ1, KCNH2, SCN5A) suffisent désormais à poser le diagnostic.
Trois types
On distingue 3 formes selon les anomalies génétique retrouvées : SQTL de type 1 (associés à une mutation du gène KCNQ1 ; 40 - 55 % des cas), SQTL 2 (mutation de KCNH2, 30 - 45 % des cas) et SQTL 3 (mutation du gène SCN5A ; 5 - 10 % des cas). Ces types de SQTL diffèrent aussi par les anomalies observées à l’ECG et les modes de déclenchement des symptômes (arrêt cardiaque ou syncope) : respectivement, on les retrouve plutôt à l’effort (SQTL 1), dans un contexte émotionnel/au réveil (SQTL 2), au repos/dans le sommeil (SQTL 3).
Plus précisément, dans le SQTL 1, ce sont surtout les garçons pré-pubertaires qui sont symptomatiques, typiquement lors d’un effort physique ou sportif, ou lors d’un stress émotionnel. Dans le SQTL 2, ce sont plutôt les filles après la puberté ou en post-partum (en particulier les 9 premiers mois post-accouchement) qui sont symptomatiques, parfois après un stress auditif ou émotionnel pendant le sommeil (réveil brutal typiquement). Enfin, dans le SQTL 3, les symptômes surviennent plutôt après la puberté, souvent au repos ou durant le sommeil, en présence d’une bradycardie relative et en l’absence de stress.
Diagnostics différentiels (dont SQTL acquis)
La prolongation acquise de l’intervalle QT est due le plus souvent à des médicaments, comme certains antibiotiques (macrolides, fluoroquinolones), antidépresseurs : citalopram, escitalopram) et antipsychotiques (halopéridol, rispéridone). Une liste en anglais régulièrement actualisée est disponible gratuitement sur inscription sur le site https ://www.crediblemeds.org/ [liste de 2017 sans inscription ici]).
Le SQTL acquis peut aussi s’expliquer par un trouble électrolytique, une hypokaliémie, une hypomagnésémie voire une hypocalcémie, une bradycardie majeure, une cardiopathie (syndrome de Takotsubo, infarctus récent du myocarde), certains AVC, les hémorragies méningées, et plus rarement la prise de cocaïne ou de méthadone.
On peut aussi rencontrer un allongement de l’intervalle QT chez les athlètes pratiquant de façon intensive (plus de 10 heures par semaine). Chez eux, un QTc < 500 ms en l’absence de symptômes ou de maladie familiale est peu susceptible de représenter un SQTL.
Enfin, il ne faut pas confondre une syncope due au SQTL avec une crise épileptique familiale : une crise d’épilepsie pendant l’effort, l’excitation ou le sommeil et, en règle générale, n’ayant pas fait sa preuve neurologique, doit faire envisager un SQTL.
Traitement
L’objectif est de prévenir les syncopes. En France, la prise en charge des SQTL peut se faire en ALD au titre des troubles du rythme graves : troubles du rythme ventriculaire pouvant entraîner une instabilité hémodynamique et une mort subite cardiaque (ALD5).
Les médicaments susceptibles d’allonger l’intervalle QT sont formellement contre-indiqués.
Le risque d’accident rythmique grave (qui dépend du sexe, de l’âge, du QTc et de la forme génétique) est significativement diminué sous bêtabloquant. Ce traitement est de préférence le nadolol (1 - 1,5 mg/kg/j), ou le propranolol (2 - 3,5 mg/kg/j), recommandé pour tous les patients avec un diagnostic clinique de SQTL. Attention : contrairement aux autres bêtabloquants, le sotalol est strictement contre-indiqué, car il allonge l’intervalle QT. Le métoprolol ne doit pas être utilisé.
En cas de SQTL 3, c’est un traitement à la mexilétine qui est recommandé (8 mg/kg/j). Par ailleurs, des données récentes montrent que 70 % des SQTL 2 répondent aussi à cette molécule : chez ces patients, un traitement d’épreuve peut être réalisé avant une éventuelle prescription : administration orale de 6 - 8 mg/kg de mexilétine (l’efficacité est validée si le QTc diminue de plus de 40 ms dans les 2 heures post-administration).
Pour les enfants, il faut demander un avis spécialisé (dans un centre de référence ou de compétence) afin de préciser les modalités de suivi et le moment opportun pour instaurer le traitement.
Si des syncopes surviennent malgré un traitement bêtabloquant à pleine dose, la dénervation sympathique cardiaque gauche peut être considérée et proposée sans hésitation. Elle peut être proposée chez les patients symptomatiques ne tolérant pas de bêtabloquants, ou chez qui ils sont contre-indiqués.
Un défibrillateur automatique implantable (DAI) est recommandé chez les patients qui ont :
- survécu à un arrêt cardiaque (en association à un traitement bêtabloquant) ;
- des syncopes malgré un traitement bêtabloquant à pleine dose, si la mexilétine et la dénervation sympathique cardiaque gauche ne sont pas disponibles ;
- des syncopes malgré un traitement bêtabloquant à pleine dose et une dénervation sympathique cardiaque gauche ;
- des syncopes mais ne tolèrent pas les médicaments (bétabloquants ou méxilétine).
Enfin, l’implantation d’un DAI peut être considérée chez les patients asymptomatiques ayant un profil à haut risque selon le nouveau calculateur 1 - 2 - 3 LQTS risk, en plus d’un traitement médicamenteux adapté à leur génotype (bétabloquants ou méxilétine).
Sport et autres mesures préventives
Les sports de compétition sont contre-indiqués. Les sports de loisirs sont autorisés sous réserve d’absence de symptômes, de niveau d’effort modéré et de contrôle du traitement bêtabloquant à l’épreuve d’effort (fréquence cardiaque maximale < 150/bpm ou < 70 % de la fréquence maximale théorique, avec FMT = 220 - l’âge).
L’autorisation de pratiquer la natation et les sports aquatiques est une décision très délicate en cas de SQTL 1. De même, l’évitement des situations de stress sonore est à discuter pour les SQTL 2. La prévention des désordres métaboliques (hypokaliémie, hypomagnésémie) est nécessaire dans tous les cas.
La prise en charge psychologique est essentielle. Il faut expliquer au patient :
- la nécessité légale d’informer (lui-même ou via le médecin après autorisation écrite) les membres de sa famille (parents, fratrie et descendance) de l’existence du syndrome du QT long ;
- qu’il s’agit d’une affection risquant d’être transmise une fois sur deux à chaque grossesse, quel que soit le sexe.
HAS. Syndrome du QT long – synthèse du protocole national de diagnostic et de soins (PNDS) à destination du médecin traitant. 14 octobre 2021.
HAS. Syndrome du QT long – protocole national de diagnostic et de soins. 14 novembre 2021.
Pour en savoir plus :
Nobile C. Syndrome du QT long : une fiche pour le MG ? Rev Prat (en ligne) 24 mai 2022.
Minh S, Paret S. Vivre avec un syndrome du QT long. Rev Prat 2016;66(4):405-6.
Denjoy I, Lupoglazoff JM, Extramiana et al. Le syndrome du QT long congénital : une cause inhabituelle de mort subite. Réalités Cardio 22 octobre 2010.
Zeppenfeld K, Tfelt-Hansen J, de Riva M, et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: Developed by the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC) Endorsed by the Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J 2022;43(40):3997-4126.
Groffen AJ, Bikker H, Christiaans I. Long QT Syndrome Overview. GeneReviews 21 mars 2024
Wilde AAM, Amin AS, Postema PG. Diagnosis, management and therapeutic strategies for congenital long QT syndrome. BMJ Heart 16 février 2022.
Cardiogen. Syndrome du QT long.