Le syndrome d’activation macrophagique (SAM), ou lymphohistiocytose hémophagocytaire (LH), est causé par une dérégulation de la réponse immunitaire, entraînant une inflammation incontrôlée et inefficace.1 Le terme SAM est ici utilisé car il s’agit du terme consacré en littérature francophone, mais dans la littérature anglophone ce terme est employé pour désigner spécifiquement la LH en contexte de maladie auto-immune.
Le SAM n’est pas une maladie en soi mais plutôt la conséquence d’une incapacité génétique ou acquise du système immunitaire à faire face à une stimulation qui, dans la plupart des cas, est un agent infectieux ou un cancer. Il est caractérisé par une hyperactivation des lymphocytes T CD8 et des macrophages, entraînant une « tempête » cytokinique, une hémophagocytose (phagocytose des cellules sanguines par suractivation des macrophages), une fièvre élevée persistante, une hépatosplénomégalie, des cytopénies, une hyperferritinémie, une coagulopathie et une hypertriglycéridémie. Sans traitement, l’évolution se fait vers une défaillance multiviscérale.1,2 La LH est classée comme primaire d’origine génétique (LHp) ou secondaire acquise (LHs).
Les LHp sont des déficits immunitaires autosomiques récessifs le plus souvent liés à l’inactivation de gènes codant pour des protéines cruciales pour la cytotoxicité des lymphocytes T et des cellules natural killer (NK).2 Ces formes touchent principalement les nourrissons et les jeunes enfants, mais des révélations tardives sont de plus en plus fréquemment rapportées chez l’adulte.1 Les LHp restent des formes très rares.
Les LHs sont les plus fréquentes, et on retrouve souvent un « facteur prédisposant » entraînant une dérégulation du système immunitaire comme une néoplasie, une immunodépression, une maladie auto-immune/auto-inflammatoire et/ou un « trigger » (déclencheur) le plus souvent infectieux.3
Le SAM est une pathologie grave, avec une mortalité globale particulièrement élevée, de 42 % en moyenne (avec des variations de 20 à 70 % selon le terrain et la durée de suivi évaluée),4 nécessitant une prise en charge diagnostique et thérapeutique rapide.
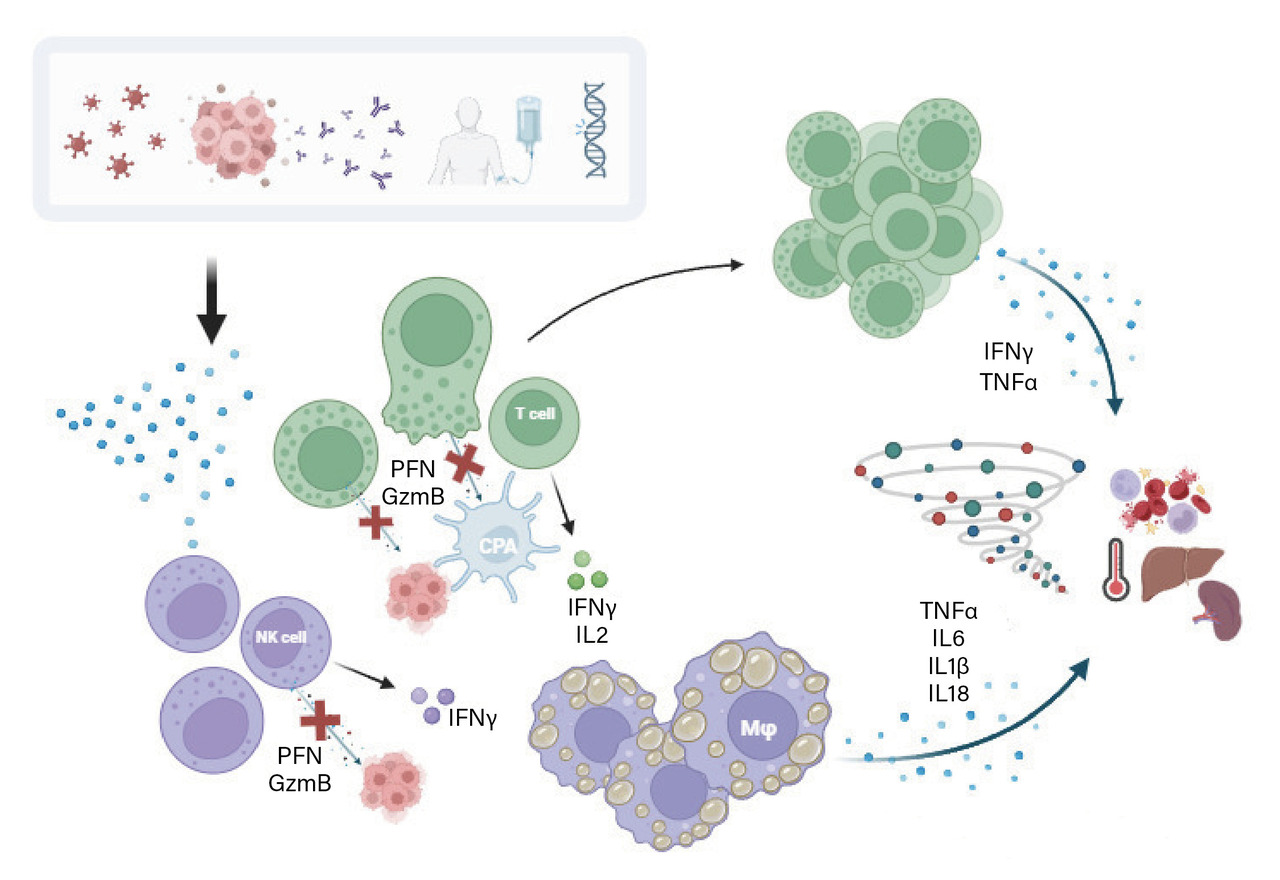
Physiopathologie : défaut de cytotoxicité des cellules NK et des lymphocytes T CD8
Même si les causes diffèrent, les SAM partagent une physiopathologie commune : le dénominateur commun est le défaut de cytotoxicité des cellules NK (immunité innée) et des lymphocytes T (LT) CD8 (immunité adaptative) [fig. 1]. Ce défaut entraîne une surstimulation du système immunitaire, conduisant à une tempête de cytokines pro-inflammatoires et à une activation T incontrôlée responsables des manifestations cliniques et biologiques du SAM.2 Les LT CD8 activés libèrent de l’interféron (IFN)-γ, qui stimule les macrophages induisant la production de cytokines pro-inflammatoires comme le TNF-α, l’interleukine (IL)- 1, l’IL- 6, l’IL- 18 et des chimiokines. Ces niveaux élevés de cytokines pro-inflammatoires sont responsables de la fièvre, inhibent l’hématopoïèse et la lipoprotéine lipase, provoquant des cytopénies et une hypertriglycéridémie. L’activation des macrophages conduit à des niveaux accrus d’activateurs du plasminogène entraînant une hyperfibrinolyse et à des niveaux élevés de ferritine.2 Le défaut de cytotoxicité peut être permanent dans les formes génétiques ou transitoire dans les formes secondaires. Les facteurs prédisposants secondaires comprennent les déficits immunitaires acquis tels que l’infection par le VIH et les immunosuppresseurs, mais aussi les tumeurs malignes, plus rarement des maladies auto-inflammatoires et auto-immunes.
Étiologie des SAM
Formes primitives
La LHp est rare, avec une incidence estimée à environ 1 cas sur 50 000 à 100 000 naissances vivantes. L’inactivation de plusieurs gènes impliqués dans la cytotoxicité a été décrite (PRF1, MUNC13 - 4, STXBP2, STX11, SH2D1A, BIRC4). L’inactivation complète de l’un de ces gènes conduit le plus souvent à des formes mortelles, avec une survie médiane inférieure à deux mois après le diagnostic en l’absence de traitement et qui se manifeste généralement pendant la petite enfance. Les antécédents familiaux sont souvent absents du fait du mode de transmission récessif.1 La LHp est traditionnellement considérée comme une maladie pédiatrique, mais l’évaluation génétique systématique d’adolescents et d’adultes atteints de SAM a permis d’identifier des mutations hypomorphes de ces gènes chez 7 à 14 % des patients, ce qui suggère que la LHp à révélation tardive est plus fréquente qu’on ne le pensait.1
Formes secondaires
Chez l’adulte, les SAM sont principalement des LHs. Les principales causes de LHs sont les infections et les tumeurs malignes. Les maladies auto-inflammatoires et auto-immunes sont beaucoup plus rares. Les études rétrospectives menées chez l’adulte mettent en évidence une prédominance de SAM associés à des néoplasies (de 50 à 60 %), avec une majorité d’hémopathies malignes, suivie par les causes infectieuses (de 25 à 35 %), puis les maladies auto-immunes (de 3 à 8 %) et rarement les tumeurs solides.4,5 Le SAM chez l’adulte est souvent multifactoriel, avec un terrain d’immunodépression et un ou plusieurs facteurs déclenchants ; la recherche étiologique doit donc être la plus large possible.
Tumeurs malignes
Un SAM peut survenir chez jusqu’à 1 % des patients atteints de tumeurs malignes.6 On distingue le SAM déclenché par la tumeur maligne, dans ce cas il est présent au diagnostic ou à la rechute, et le SAM survenant pendant la chimiothérapie, ce dernier étant souvent déclenché par des infections liées à l’immunodépression secondaire ou par l’utilisation de certaines thérapeutiques.6 Les SAM sont le plus souvent décrits au cours des lymphomes malins non hodgkiniens (LMNH) T et NK (35 %), des LMNH B diffus à grandes cellules (32 %), des lymphomes de Hodgkin (6 %) et des leucémies (6 %).6
Infections
Les virus, en particulier ceux de la famille des Herpesviridae, sont de puissants inducteurs de SAM. Le virus d’Epstein-Barr (EBV) est le trigger le plus fréquent et le plus puissant de SAM, avec une prédisposition génétique particulière en Asie. Des SAM induits par HHV- 8 sont également décrits dans la littérature, la plupart du temps dans le cadre d’un sarcome de Kaposi ou d’une maladie de Castleman multicentrique et chez des patients immunodéprimés (sida, transplantation d’organe).7 Enfin, le VIH, surtout en présence d’autres infections opportunistes ou de néoplasie, a été décrit comme associé à la survenue de SAM.8
Outre les virus, d’autres agents pathogènes ont été impliqués dans le déclenchement de SAM, notamment les protozoaires, les bactéries pyogènes, les mycobactéries et les champignons. Les pathogènes intracellulaires semblent avoir une propension particulière à déclencher des SAM, notamment la leishmaniose viscérale et la tuberculose, qui sont les triggers infectieux non viraux les plus fréquents.7
Maladies auto-immunes/inflammatoires
Le SAM est une complication potentiellement mortelle des pathologies inflammatoires systémiques. Il survient plus souvent au cours de la maladie de Still de l’adulte, de l’arthrite juvénile idiopathique systémique et du lupus érythémateux systémique, mais a également été décrit dans d’autres maladies auto-immunes.9 La prévalence estimée du SAM dans ces populations varie de 1 à 9 % au cours du lupus systémique et jusqu’à 10 à 15 % au cours de la maladie de Still de l’adulte2 dont il peut être la manifestation inaugurale.
Diagnostic : rechercher les facteurs déclenchants et prédisposants
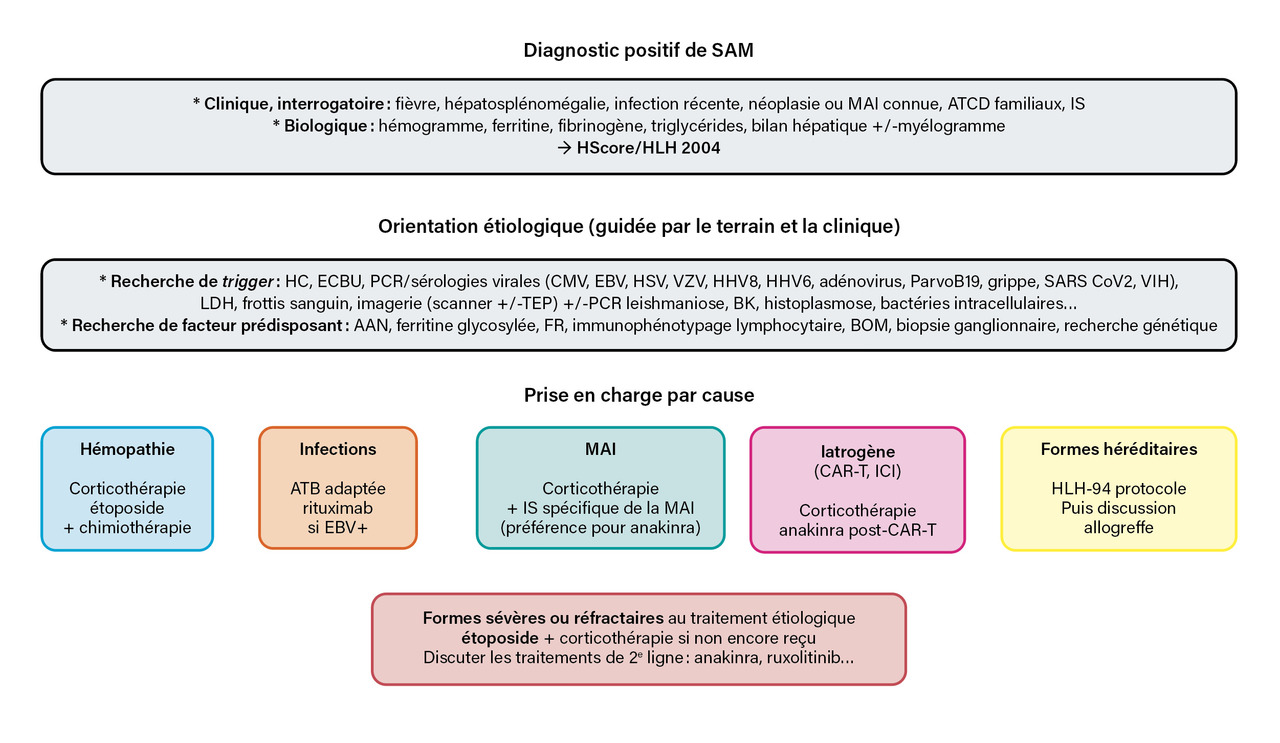
Trois grandes questions diagnostiques doivent être abordées chez un patient présentant une suspicion de SAM (fig. 2) : ce patient est-il atteint de SAM ? Existe-t-il un facteur déclenchant aigu ? Quel est le facteur prédisposant ?
Présentation clinique, biologique et histologique évocatrice
Aucune manifestation clinique ni biologique n’est spécifique d’un SAM, mais c’est leur association et leur évolutivité au cours du temps qui doit mener à la suspicion diagnostique. Les signes cardinaux sont une fièvre prolongée et des cytopénies. Une hépatosplénomégalie est présente dans environ deux tiers des cas, des manifestations plus rares sont possibles (pulmonaires, neurologiques…). L’hyperferritinémie est également une caractéristique essentielle car quasi constante avec, dans certaines études, un seuil supérieur à 10 000 µg/l associé à un diagnostic de SAM, avec une spécificité de 96 % et une sensibilité de 90 %.2 On observe aussi fréquemment une hypertriglycéridémie, une coagulopathie avec hypofibrinogénémie (mais la coagulation intravasculaire disséminée [CIVD] est rare), une dysfonction hépatique. D’autres anomalies biologiques ont été également décrites, comme une faible activité des cellules NK et des taux élevés de récepteurs solubles de l’interleukine 2 (sIL- 2r ou sCD25), mais elles sont moins accessibles en pratique clinique.10 Les anomalies cytopathologiques comprennent une accumulation de lymphocytes T activés et de macrophages dans les tissus, mais surtout une image d’hémophagocytose (macrophage phagocytant des cellules sanguines) au niveau de la moelle osseuse et visible également dans le foie et les ganglions. Cependant, l’hémophagocytose peut ne pas être présente aux stades initiaux et n’est pas spécifique du SAM.6 Un prélèvement initial négatif ne doit donc pas retarder le diagnostic ni le début du traitement si les autres paramètres sont évocateurs de SAM. Dans une série de 162 patients atteints de SAM, l’hémophagocytose était observée chez environ 70 % des patients dont le diagnostic de SAM a été confirmé ; mais elle était également observée chez environ 40 % des patients avec un tableau clinique similaire mais ne répondant pas aux autres critères de SAM.5
Scores diagnostiques
Le diagnostic de SAM repose sur l’association d’un ensemble de critères cliniques, biologiques et cytologiques car aucun élément pris isolément n’est spécifique (tableau). En 1991, l’Histiocyte Society a présenté les premières recommandations d’aide au diagnostic du SAM, fondées sur des éléments cliniques, biologiques et histopathologiques, qui ont été actualisées en 2004.
Les recommandations HLH- 2004 (Hemophagocytic lymphohistiocytosis) comprennent un ensemble de huit critères : fièvre, splénomégalie, cytopénie (touchant au moins deux lignées), hypertriglycéridémie et/ou hypofibrinogénémie, hémophagocytose, activité faible ou absente des cellules NK, hyperferritinémie et taux élevé de sCD25.10 Au total, cinq des huit critères doivent être remplis, à l’exception des patients dont le diagnostic moléculaire permet de retenir le diagnostic de LHp. Cependant, les recommandations de 1991 et de 2004 ont été élaborées à partir de populations pédiatriques atteintes de LHp, et le diagnostic moléculaire, le dosage du sCD25 et l’étude de la fonction des NK sont rarement réalisés en pratique clinique chez l’adulte.
Pour aider au diagnostic de LHs, le HScore a été créé en 2014, intégrant neuf caractéristiques cliniques et biologiques du SAM (tableau). Il est utilisable par les cliniciens pour établir un score de probabilité diagnostique de SAM et peut être calculé en ligne (http ://saintantoine.aphp.fr/score/).11 Un patient est défini comme ayant un SAM si le HScore est de 169 ou plus, ce qui permet de classer avec précision 90 % des patients (sensibilité : 93 % ; spécificité : 86 %).Enfin, des critères ont été publiés en pédiatrie pour distinguer une poussée d’arthrite juvénile idiopathique d’un SAM.12
Recherche des facteurs prédisposants et déclenchants
La recherche de facteurs prédisposants (infection VIH, autres causes d’immunosuppression) et déclenchants (infection, tumeur) doit se faire en parallèle de la mise en place d’une thérapeutique et ne doit pas retarder la prise en charge spécifique du SAM. Les triggers viraux, en particulier les virus du groupe herpès, sont les plus fréquents et doivent être activement recherchés par polymerase chain reaction (PCR) et sérologies dédiées. D’autres causes infectieuses, telles que des agents bactériens, protozoaires et fongiques, doivent également être envisagées. La leishmaniose, la rickettsiose et la tuberculose notamment doivent être recherchées en cas d’origine de zones endémiques.1 Les histoplasmoses et toxoplasmoses disséminées se voient chez des patients immunodéprimés.
La probabilité d’une tumeur maligne sous-jacente augmente avec l’âge. Un lymphome doit être recherché en priorité, surtout en l’absence de cause infectieuse ou auto-immune facilement identifiée. Le bilan comprend un frottis sanguin, des explorations médullaires (myélogramme et biopsie ostéomédullaire) et un bilan d’imagerie avec un scanner pouvant être complété par une tomographie par émission de positons (TEP) à la recherche de cible à biopsier.
Depuis quelques années, les traitements des cancers par eux-mêmes sont des causes de SAM ; c’est le cas des inhibiteurs des points de contrôle de la réponse immunitaire et des thérapies par cellules T à récepteur d’antigène chimérique (CAR-T).13,14 Dans les deux cas, le système immunitaire est « boosté » contre les cellules cancéreuses, ce qui peut entraîner son activation excessive.
La recherche d’une maladie auto-immune et auto-inflammatoire est guidée par les manifestations cliniques (notamment la présence d’arthralgie ou d’arthrite, de lésions cutanées) et certaines caractéristiques biologiques (présence d’auto-anticorps antinucléaires/anti-ADN pour le lupus ou taux de neutrophiles passant d’un niveau initialement très élevé à des valeurs normales ou réduites pour la maladie de Still).12
Les caractéristiques cliniques devant faire suspecter une LHp chez l’adulte comprennent la présence d’antécédents familiaux, une consanguinité, la survenue de rechutes, un albinisme partiel, l’association avec des troubles digestifs ou une réponse altérée ou aberrante à l’EBV.1
Diagnostics différentiels
De nombreuses affections peuvent donner lieu à un tableau proche du SAM en cas d’activation intense du système immunitaire, principalement au cours d’infections sévères ou d’hémopathie maligne. Il est donc essentiel de déterminer si la combinaison, l’étendue et l’évolution des anomalies cliniques et biologiques sont inhabituelles, inattendues ou inexpliquées pour évoquer le diagnostic de SAM.1
Éléments pronostiques
Une large étude, décrivant 162 patients atteints de SAM quelle qu’en soit la cause, a rapporté une mortalité globale de 42 %.5 La mortalité était généralement plus faible chez les patients atteints de maladies auto-immunes, suivis par les patients atteints d’un SAM idiopathique ou associé à une infection. Les néoplasies, en particulier le SAM associé au lymphome, constituent un facteur pronostique défavorable.6 Parmi les autres facteurs pronostiques défavorables, on peut citer un âge supérieur à 30 ans, une ferritinémie très élevée, une thrombopénie marquée, une hypoalbuminémie, le sexe masculin et l’absence de traitement par étoposide.15
Double objectif du traitement
L’objectif de la prise en charge thérapeutique du SAM est à la fois de traiter tout facteur déclenchant et de contrôler l’hyperactivité du système immunitaire (fig. 2).
Si une tumeur maligne ou une infection sont identifiées, un traitement spécifique doit être mis en place le plus rapidement possible. Ces mesures sont parfois suffisantes pour mettre fin à la dysrégulation immunitaire. Ainsi, le traitement de l’infection sous-jacente est associé à une guérison chez 60 à 70 % des patients et ne nécessite généralement pas de traitement complémentaire.7 Un traitement immunosuppresseur supplémentaire doit être mis en place d’emblée dans les cas graves, ou ajouté rapidement chez les patients ne répondant pas au traitement spécifique de la maladie.9
Le traitement immunosuppresseur repose largement sur la corticothérapie, les schémas commençant souvent par de fortes doses (jusqu’à 30 mg/kg/j de méthylprednisolone, au maximum 1 g/j). Les patients avec un SAM modéré ou débutant sont susceptibles de répondre à une corticothérapie systémique isolée sans agent cytostatique.
Dans les SAM sévères, en revanche, le pronostic a été drastiquement amélioré par l’utilisation de l’étoposide. Il est désormais considéré comme le traitement de référence des SAM graves et des SAM associés à un lymphome où son ajout directement au protocole de chimiothérapie permet d’améliorer la survie globale.6 Il est utilisé à une dose de 50 à 100 mg/m2 par semaine adaptée à l’âge et pour une durée réévaluée de façon hebdomadaire.1,10 Il agit en induisant sélectivement une forte déplétion des cellules T activées et une suppression de la production de cytokines inflammatoires. Dans les cas réfractaires, un immunosuppresseur supplémentaire doit être instauré, il peut reposer sur l’utilisation de certaines biothérapies. L’anakinra (antagoniste du récepteur de l’IL- 1), souvent préféré en raison de sa rapidité d’action et de son profil d’effets indésirables acceptable, et le ruxolitinib (inhibiteur de Janus kinases 1 et 2) sont les agents facilement disponibles en deuxième intention.1
Les patients avec une forme génétique nécessitent le plus souvent la réalisation d’une allogreffe de cellules souches hématopoïétiques.
La réponse au traitement est déterminée par l’évolution clinique et biologique : résolution de la fièvre, normalisation de la ferritinémie, de la fonction hépatique et du fibrinogène. Le monitoring du taux de ferritine sérique pendant et après le traitement semble être particulièrement corrélé avec l’activité de la maladie. L’évolution des cytopénies, quant à elle, peut être perturbée en cas d’utilisation d’étoposide en raison de son effet myélotoxique.
Hyperactivité du système immunitaire à traiter rapidement
Le SAM est une réaction inflammatoire incontrôlée et potentiellement mortelle causée par une immunodéficience et/ou une pathologie néoplasique, infectieuse ou auto-immune. Le diagnostic précoce et le traitement rapide de tout facteur déclenchant et de l’hyperactivité du système immunitaire constituent la base de la prise en charge.
2. Carter SJ, Tattersall RS, Ramanan AV. Macrophage activation syndrome in adults: Recent advances in pathophysiology, diagnosis and treatment. Rheumatology (Oxford) 2019;58:5-17.
3. Ramos-Casals M, Brito-Zerón P, López-Guillermo A, et al. Adult haemophagocytic syndrome. The Lancet 2014;383:1503-16.
4. Arca M, Fardet L, Galicier L, et al. Prognostic factors of early death in a cohort of 162 adult haemophagocytic syndrome: Impact of triggering disease and early treatment with etoposide. Br J Haematol 2015;168:63-8.
5. Riviere S, Galicier L, Coppo P, et al. Reactive hemophagocytic syndrome in adults: A retrospective analysis of 162 patients. Am J Med 2014;127:1118-25.
6. Lehmberg K, Nichols KE, Henter JI, et al. Consensus recommendations for the diagnosis and management of hemophagocytic lymphohistiocytosis associated with malignancies. Haematologica 2015;100:997-1004.
7. Rouphael NG, Talati NJ, Vaughan C, et al. Infections associated with haemophagocytic syndrome. Lancet Infect Dis 2007;7:814-22.
8. Fardet L, Lambotte O, Meynard JL, et al. Reactive haemophagocytic syndrome in 58 HIV-1-infected patients: Clinical features, underlying diseases and prognosis. AIDS 2010;24:1299-306.
9. Schram AM, Berliner N. How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood 2015;125:2908-14.
10. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48:124-31.
11. Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis & Rheum 2014;66(9):2613-20.
12. Shakoory B, Geerlinks A, Wilejto M, et al. The 2022 EULAR/ACR points to consider at the early stages of diagnosis and management of suspected haemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS). Ann Rheum Dis 2023;82:1271-85.
13. Zhai C, Jin X, You L, et al. Hemophagocytic lymphohistiocytosis following pembrolizumab and bevacizumab combination therapy for cervical cancer: A case report and systematic review. BMC Geriatr 2024;24:32.
14. Hines MR, Knight TE, McNerney KO, et al. Immune effector cell-associated hemophagocytic lymphohistiocytosis-like syndrome. Transplant Cell Ther 2023;29:438.e1-438.e16.
15. Hayden A, Park S, Giustini D, et al. Hemophagocytic syndromes (HPSs) including hemophagocytic lymphohistiocytosis (HLH) in adults: A systematic scoping review. Blood Rev 2016;30:411-20.