Le syndrome de Cushing regroupe l’ensemble des manifestations induites par une exposition chronique à un excès de glucocorticoïdes, qu’il soit d’origine endogène ou exogène ; seul le syndrome de Cushing d’origine endogène est traité dans ce dossier. Il est responsable de la survenue de nombreux symptômes cliniques (vergetures, amyotrophie, érythrose du visage, fragilité cutanée, troubles neurocognitifs, troubles de l’humeur…)1 et de complications comme l’hypertension artérielle, le diabète, la dyslipidémie, l’obésité, les pathologies coronariennes, les troubles psychiques ou encore l’ostéoporose.2 Ces complications contribuent à une surmortalité. La précocité du diagnostic et du contrôle de l’excès de glucocorticoïdes est déterminante pour limiter les effets délétères et les séquelles du syndrome de Cushing.
Épidémiologie et formes étiologiques
Le syndrome de Cushing est une pathologie rare, avec une incidence annuelle de l’ordre de 1,2 à 5 par million d’habitants et par an. L’âge moyen d’apparition est de 41,4 ans et il concerne principalement les femmes, avec un ratio femme/homme de 3 :1.3
Deux situations sont possibles :
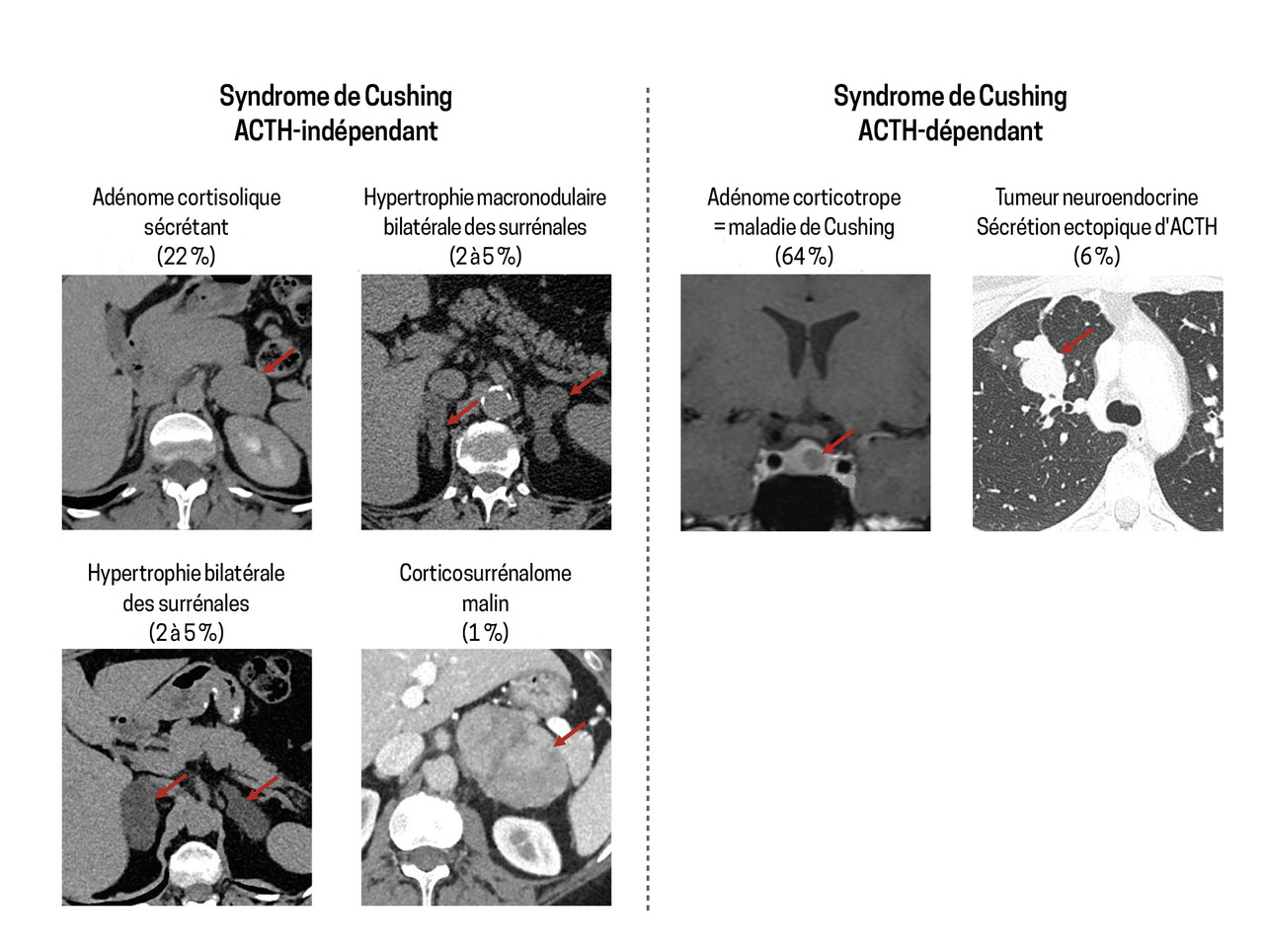
- dans 70 à 80 % des cas, la sécrétion accrue de cortisol par les glandes surrénales est due à une sécrétion excessive et inappropriée de l’hormone corticotrope (ACTH) ; on parle de syndrome de Cushing ACTH-dépendant ; dans environ 85 à 90 % de ces cas, l’ACTH est sécrétée par une tumeur corticotrope hypophysaire : c’est la maladie de Cushing. Dans 10 à 15 % de ces cas, l’ACTH est d’origine ectopique, produite par une tumeur neuroendocrine non hypophysaire (pulmonaire, pancréatique, etc.) ;
- dans 20 à 30 % des cas, la sécrétion excessive de cortisol provient des glandes surrénales et inhibe la sécrétion d’ACTH hypophysaire par un effet de rétro-contrôle négatif ; on parle de syndrome de Cushing ACTH-indépendant. Dans ces cas, l’hypercortisolisme est lié à un adénome corticosurrénalien unilatéral (environ 70 % des cas), à un cancer corticosurrénalien (30 %) ou à une hypertrophie nodulaire bilatérale des surrénales (environ 2 %).3
Des symptômes le plus souvent non spécifiques
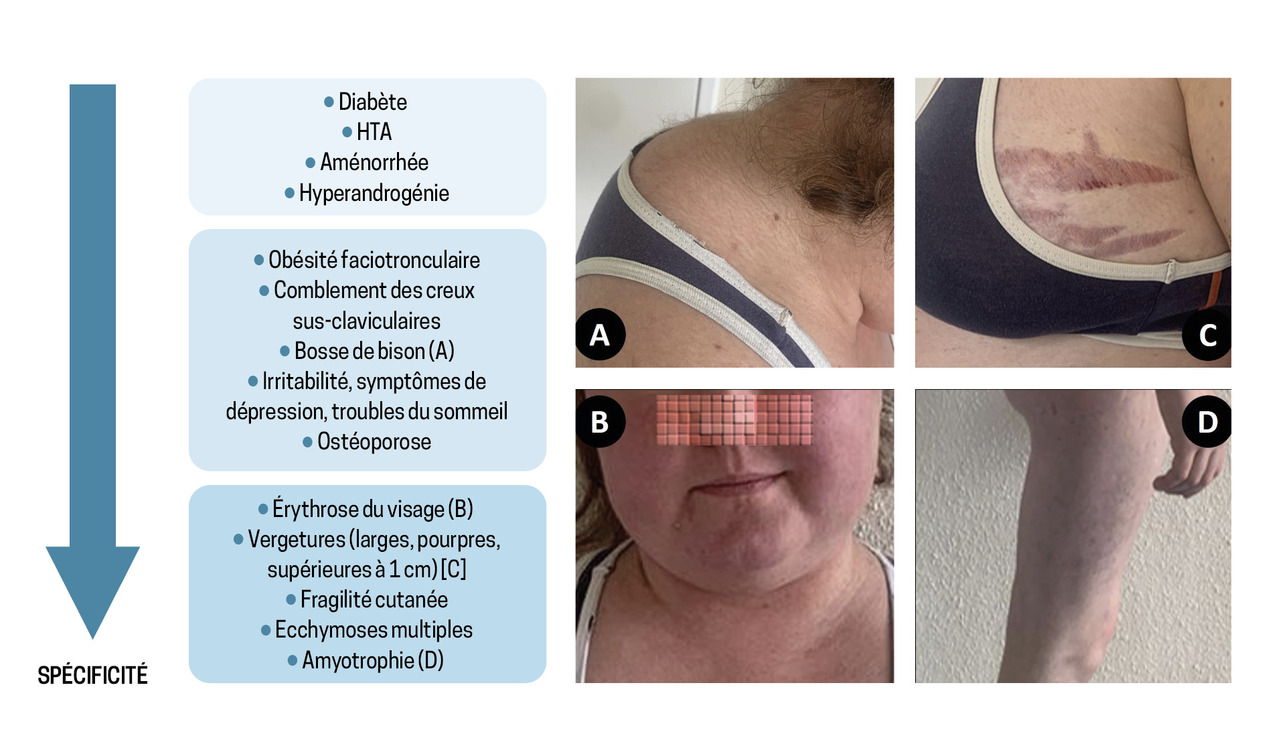
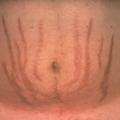
La présentation clinique du syndrome de Cushing est très variable selon l’intensité, la durée d’exposition et la susceptibilité individuelle à l’excès de cortisol (fig. 1).
L’une des difficultés diagnostiques tient au fait que les symptômes fréquemment retrouvés – tels que la prise pondérale, le diabète sucré, l’hypertension artérielle, les symptômes de dépression – sont également très fréquents dans la population générale, et donc peu discriminants.
À l’inverse, les symptômes plus spécifiques (vergetures pourpres, myopathie proximale, fragilité cutanée), reflétant les effets cataboliques de l’excès de cortisol et dont la présence augmente la probabilité diagnostique, ne sont pas toujours présents et sont souvent associés à un syndrome de Cushing intense ou ayant débuté depuis plusieurs années. L’aggravation de la symptomatologie dans le temps, la survenue de comorbidités à un âge jeune ou en l’absence de facteur de risque associé, et leur évolution inhabituelle dans le temps sont autant d’éléments devant faire penser à un syndrome de Cushing.1,4
Les recommandations de la Société française d’endocrinologie4 suggèrent de réaliser un dépistage biologique du syndrome de Cushing dans les cas suivants :
- répartition faciotronculaire des graisses en présence d’un excès pondéral modéré ou au moins une manifestation clinique « spécifique » ;
- présence d’une ou plusieurs comorbidités mais dont la prévalence est inhabituelle pour l’âge (hypertension artérielle avant 40 ans, ostéoporose chez l’homme avant 50 ans, etc.) ou dont l’évolution est inhabituelle lors d’un traitement conventionnel ;
- découverte d’un incidentalome surrénalien compatible avec un adénome, un adénome hypophysaire ou toute tumeur pouvant induire un syndrome de Cushing par sécrétion ectopique d’ACTH.
À l’inverse, il n’est pas recommandé de réaliser un dépistage biologique systématique chez les patients diabétiques, hypertendus ou obèses asymptomatiques, puisque dans ces populations, la prévalence de ce syndrome est très faible (inférieure à 2 %), et la probabilité d’obtenir un faux positif au test de dépistage (environ 10 %) est largement supérieure à celle d’obtenir un vrai positif.4
Modalités diagnostiques et d’exploration
En dehors des cas cliniquement évidents, si un syndrome de Cushing est suspecté, le diagnostic passe par quatre grandes étapes :
- dépistage biologique de l’hypercortisolisme en ambulatoire à l’aide d’un test de dépistage ;
- confirmation biologique de l’hypercortisolisme en centre expert ;
- détermination du caractère ACTH-dépendant ou indépendant du syndrome ;
- diagnostic étiologique du syndrome de Cushing.
Toute pathologie intercurrente (infection, traumatisme, etc.) ainsi que l’alcoolisme, la dépression et autres maladies psychiatriques activent physiologiquement l’axe hypothalamo-hypophysaire et augmentent la sécrétion de cortisol, pouvant simuler biologiquement un syndrome de Cushing ACTH-dépendant d’intensité faible à modérée. Il est donc important de réaliser les explorations biologiques en dehors de ces contextes ou d’en tenir compte dans l’interprétation des résultats.
Dépistage du syndrome de Cushing en ambulatoire
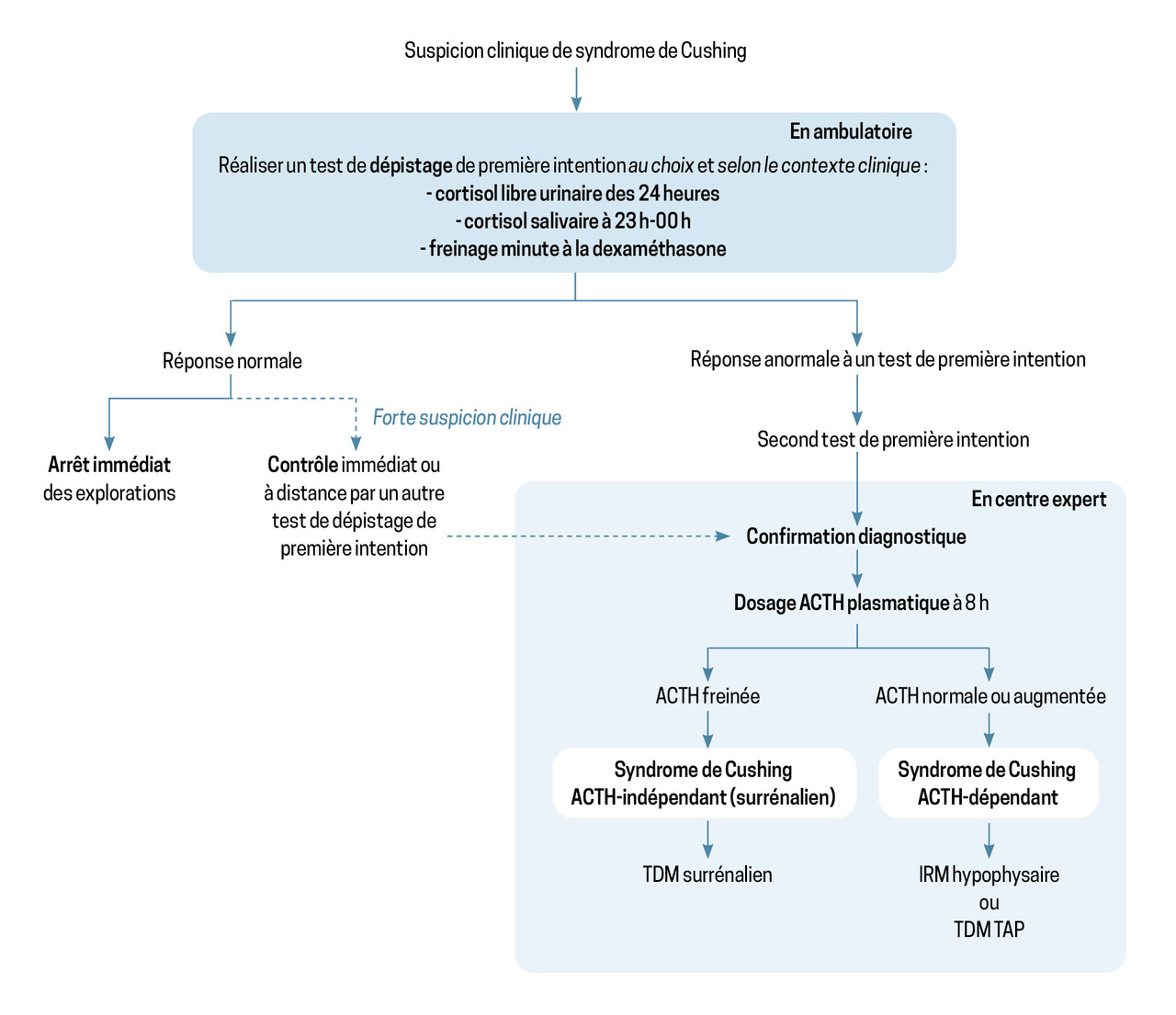
En cas de suspicion de syndrome de Cushing, il est recommandé de réaliser en ambulatoire l’un des tests de dépistage de première intention, parmi les suivants :
- test de freinage minute à la dexaméthasone (overnight dexamethasone suppression test [ODST]) ;
- cortisol libre urinaire (CLU) des vingt-quatre heures et créatininurie ;
- cortisol salivaire vespéral (late-night salivary cortisol).1,4
Les performances de ces trois tests sont comparables pour le diagnostic de syndrome de Cushing (sensibilité de 94 à 96 % ; spécificité de 90 à 93 %)5, mais ils doivent être choisis en fonction des caractéristiques du patient et de l’expertise technique du laboratoire. Il est donc nécessaire de connaître les limites de chacun de ces examens.
Test de freinage minute à la dexaméthasone
L’ODST permet de vérifier si la rétroaction négative physiologique des glucocorticoïdes sur l’axe hypothalamo-hypophyso-surrénalien (HHS) est fonctionnelle. Un taux de cortisol sérique supérieur à 50 nmol/L (1,8 µg/dL) après le test offre une sensibilité diagnostique élevée (généralement 98,6 %) mais une spécificité modérée (environ 90 %). Plusieurs facteurs peuvent être responsables de faux positifs :
- la mauvaise absorption de la dexaméthasone chez les patients polymédiqués ou traités par chirurgie bariatrique ;
- l’altération du métabolisme de la dexaméthasone chez les patients traités par des inducteurs enzymatiques (carbamazépine, phénytoïne…) ;
- l’élévation de la protéine porteuse du cortisol (la transcortine, ou cortisol binding globulin [CBG]) lors des traitements par œstrogènes (pilule contraceptive, traitement hormonal de substitution de la ménopause…) qui majorent les concentrations dosées de cortisol sans modifier la concentration libre et biologiquement active du cortisol.
La présence de ces facteurs incite à préférer un autre test de dépistage.
Test du cortisol libre urinaire
Le CLU reflète la production de cortisol sur vingt-quatre heures. Puisqu’il mesure le cortisol non lié à la protéine porteuse du cortisol, le CLU n’est pas affecté par les facteurs qui modifient cette protéine porteuse. Toutefois, il est fondamental que les patients fournissent une collecte complète d’urine de vingt-quatre heures, ce qui implique d’expliquer précisément aux patients les modalités de recueil et de contrôler la complétude de ce dernier par le dosage de la créatininurie. Ce recueil n’est pas toujours réalisable et la mesure du CLU doit être proscrite, du fait de faux négatifs, dès que la clairance de la créatinine est inférieure à 60 mL/min.
Test du cortisol salivaire vespéral
La perte de la rythmicité circadienne, reflétée par l’absence d’un nadir de cortisol en milieu de nuit, est une anomalie constante dans le syndrome de Cushing. Elle peut être facilement testée chez un patient non hospitalisé par le test du cortisol salivaire vespéral (LNSC).
En pratique, les patients prélèvent un échantillon de salive deux soirs différents au moment du coucher (après 22 h). L’échantillon est stable à température ambiante ou au réfrigérateur pendant plusieurs semaines et peut être envoyé par courrier à un laboratoire de référence.
Les avantages de ce test sont la simplicité de la procédure de recueil (facilement réalisable chez l’enfant ou le patient ayant un mauvais capital veineux), l’étroite corrélation avec le cortisol plasmatique libre et l’indépendance vis-à-vis de la CBG. Le test est inadapté aux travailleurs postés ou à ceux qui ont des horaires de travail nocturne variables. Malheureusement, cet excellent test de dépistage n’est pas pris en charge par l’Assurance maladie en France et n’est, le plus souvent, disponible que dans des centres spécialisés.
Conduite à tenir après le test de première intention
Si le test de première intention est anormal, un second test de première intention doit être réalisé pour confirmer l’hypercortisolisme. La confirmation diagnostique se fait en milieu spécialisé, et, selon le profil des patients, d’autres tests (non détaillés ici) peuvent être réalisés : cycle de cortisolémie sur vingt-quatre heures, cortisol sérique à minuit, test à la dexaméthasone 2 mg/j pendant quarante-huit heures, test à la desmopressine…4
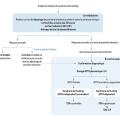
Si le test de première intention est normal, les explorations peuvent être arrêtées. La sécrétion cortisolique est néanmoins souvent fluctuante dans le syndrome de Cushing et peut épisodiquement être normale. En cas de forte suspicion clinique, la répétition d’un test peut ainsi être indiquée (fig. 2).
Diagnostic étiologique
Le diagnostic étiologique du syndrome de Cushing repose en premier lieu sur le dosage de l’ACTH plasmatique. Ce dosage ne doit être effectué qu’une fois le diagnostic positif de syndrome de Cushing certain. La fragilité de la molécule d’ACTH implique une attention particulière aux prélèvements et le recours à un laboratoire spécialisé pour son dosage.
Une concentration d’ACTH freinée est en faveur d’une origine surrénalienne à l’hypercortisolisme, alors qu’une concentration d’ACTH normale ou haute signe un syndrome de Cushing ACTH-dépendant (fig. 2). La poursuite des investigations dans cette maladie rare et complexe ne se conçoit qu’en centres experts comme ceux labellisés du réseau des maladies rares endocriniennes Firendo (www.firendo.fr).
Syndrome de Cushing ACTH-indépendant
Le diagnostic étiologique repose sur le scanner ou l’IRM surrénalienne, qui permet, dans la grande majorité des situations, de distinguer les adénomes, les corticosurrénalomes malins et les atteintes bilatérales des surrénales (hyperplasies macronodulaires ou maladies nodulaires bilatérales) [fig. 3].
L’imagerie surrénalienne ne doit être réalisée qu’après avoir constaté la suppression de la concentration d’ACTH, car il est fréquent que les patients ayant un hypercortisolisme ACTH-dépendant développent un ou plusieurs nodules surrénaliens pouvant orienter à tort vers une origine surrénalienne du syndrome de Cushing.
Syndrome de Cushing ACTH-dépendant
L’origine hypophysaire de la sécrétion d’ACTH représente plus des deux tiers des syndromes de Cushing. Dans ce cas, le syndrome est généralement d’installation progressive et d’intensité modérée.6
À l’inverse, plusieurs caractéristiques cliniques et biologiques peuvent orienter le diagnostic vers une sécrétion ectopique d’ACTH, comme l’âge avancé, le sexe masculin, et surtout l’intensité et la progression rapide des symptômes. Dans ces cas, les symptômes cataboliques (amyotrophie, fractures ostéoporotiques), l’hypertension artérielle et l’hypokaliémie sont très fréquents alors que la prise de poids est souvent absente (voire une perte pondérale est constatée). De fortes augmentations du CLU (plus de 5 fois la limite supérieure de la normale), de la cortisolémie (supérieure à 1 000 nmol/L) et des concentrations d’ACTH (supérieures à 150 pg/mL) sont également très évocatrices.
Les tests dynamiques, comme le test à la desmopressine (DDAVP), sont responsables d’environ 20 % de résultats faux positifs et négatifs et ont donc un intérêt limité.7
La détection des adénomes corticotropes repose sur l’IRM hypophysaire. Son interprétation mérite une expertise particulière car les adénomes de la maladie de Cushing sont le plus souvent des micro-adénomes (diamètre moyen de 5 mm). Ainsi, environ 20 à 30 % des patients atteints de maladie de Cushing ont une IRM normale ou non concluante, et 5 à 10 % de la population générale a de petites anomalies radiologiques compatibles avec un micro-adénome.
La recherche d’une tumeur ectopique responsable de la sécrétion d’ACTH repose en premier lieu sur la réalisation d’une tomodensitométrie (TDM) corps entier, qui retrouve une tumeur ectopique, le plus souvent pulmonaire, dans environ 70 % des cas.
En cas de doute persistant sur l’origine, d’autres examens peuvent être réalisés, comme une imagerie fonctionnelle par DOTATOC (traceur de forte affinité pour les récepteurs de type 2 de la somatostatine), voire un cathétérisme des sinus pétreux inférieurs. Le principe de cette exploration invasive – mais couramment effectuée en centre expert – est de réaliser de manière simultanée un dosage d’ACTH dans une veine périphérique et dans les premières veines de drainage de l’hypophyse. En cas de sécrétion ectopique d’ACTH, l’excès de cortisol, via un effet de rétrocontrôle négatif, supprime la sécrétion d’ACTH hypophysaire non tumorale. Ainsi, l’absence de gradient centropériphérique d’ACTH plaide en faveur d’une origine ectopique.
Divers algorithmes utilisant ces outils biologiques et radiologiques ont été proposés pour ce diagnostic, qui est considéré comme l’un des plus difficiles de l’endocrinologie.7
L’orientation clinico-biologique évoquée ci-avant apparaît fondamentale dans le choix des examens : IRM hypophysaire en premier lieu en cas de forte suspicion de maladie de Cushing ou TDM en cas d’hypercortisolisme intense (fig. 3).
Prise en charge thérapeutique
La chirurgie pour ablation de la tumeur en cause constitue le traitement de référence. Le traitement médical ne doit être envisagé qu’en deuxième intention.
Traitement chirurgical
La chirurgie de la tumeur causale (hypophysaire, surrénalienne, ectopique) est le seul traitement capable de guérir le patient. Elle doit être réalisée dans un centre expert par un chirurgien entraîné, notamment pour la neurochirurgie hypophysaire, afin d’assurer les meilleures chances de guérison, avec une morbidité minimale.8 En cas de succès, la chirurgie entraîne une insuffisance corticotrope liée à la freination chronique des cellules corticotropes « saines ». Sa durée est très variable d’un patient à l’autre – de quelques semaines à plusieurs années – et nécessite un traitement de substitution par hydrocortisone.
Dans la maladie de Cushing, la chirurgie hypophysaire par voie trans-sphénoïdale est le traitement de première intention. En centre expert, elle permet une rémission dans 70 à 80 % des cas.
En cas d’échec ou de récidive après chirurgie, plusieurs options peuvent être discutées au cas par cas : reprise de la chirurgie hypophysaire, radiothérapie hypophysaire, traitement médical ou surrénalectomie bilatérale.8
Pour les sécrétions ectopiques d’ACTH, l’urgence implique de traiter les complications de l’hyper-cortisolisme (hypokaliémie, diabète, prévention des infections, etc.) et de contrôler rapidement l’hypercortisolisme intense qui menace le pronostic vital du patient à court terme. Un traitement par inhibiteurs de la stéroïdogenèse est donc souvent introduit en première intention et précède la chirurgie curative si celle-ci est réalisable.4,8
Pour les syndromes de Cushing surrénaliens, la surrénalectomie unilatérale est le traitement de première intention dans la grande majorité des cas. Elle n’est malheureusement pas toujours réalisable en cas de corticosurrénalome (ACC) ; situation qui requiert également le recours aux chirurgiens ayant un fort volume d’activité dans cette pathologie très rare. Le traitement adjuvant par mitotane et les autres options thérapeutiques comme la chimiothérapie, la radiothérapie externe, la chimio-embolisation hépatique et la radiofréquence de localisations hépatiques ou pulmonaires se discutent au cas par cas.9
Dans les formes d’atteinte bilatérale des surrénales, la surrénalectomie bilatérale peut être indiquée, mais une surrénalectomie unilatérale peut être une alternative, notamment en cas d’asymétrie des deux glandes (résection de la plus volumineuse).
Traitement médicamenteux
Un traitement pharmacologique (tableau) peut être indiqué en cas d’échec ou d’impossibilité du traitement chirurgical, d’une maladie occulte ou métastatique, d’une récidive du syndrome de Cushing après chirurgie ou dans l’attente du traitement chirurgical.
Trois types de traitement anticortisolique peuvent être utilisés :
- les inhibiteurs de la stéroïdogenèse, qui freinent la production de cortisol par les glandes surrénales (osilodrostat, métyrapone, kétoconazole). Ils peuvent être administrés selon un schéma de titration (administration de la dose minimale efficace nécessaire au contrôle de l’hypercortisolisme sans induire d’insuffisance surrénalienne) ou, plus rarement, selon un schéma dit de « block and replace » (administration concomitante de fortes doses d’inhibiteurs de la stéroïdogenèse induisant une insuffisance surrénalienne et d’un traitement substitutif par hydrocortisone). Ils partagent des caractéristiques communes, notamment la capacité à réduire rapidement la cortisolémie, et exposent au risque de survenue d’une insuffisance surrénalienne, dont le patient doit être informé et connaître les signes d’alerte ;
- les traitements ciblant directement l’adénome hypophysaire corticotrope dans la maladie de Cushing pour réduire le sécrétion d’ACTH, et secondairement celle du cortisol. Les agonistes dopaminergiques (cabergoline) ou les analogues de la somatostatine (pasiréotide) ont toutefois une efficacité moindre (20 à 40 % de contrôle biologique) ;
- les traitements adrénolytiques (mitotane), employés principalement dans les corticosurrénalomes.
Prise en charge des complications
Le syndrome de Cushing est responsable de nombreuses complications, comme l’hypertension artérielle, le diabète sucré, l’obésité, l’ostéoporose et les fractures, les thromboses veineuses, les complications infectieuses… La disparition de ces comorbidités n’est pas toujours obtenue avec la rémission du syndrome de Cushing et leur persistance contribue, au moins en partie, au fait que les patients en rémission gardent un risque de surmortalité plus élevé que celui de la population générale.2
Des complications parfois oubliées, comme les complications neurocognitives (troubles de l’humeur, de l’attention ou de la mémoire) ou les myopathies, altèrent significativement la qualité de vie des patients, leur vie sociale et leur capacités professionnelles. Une prise en charge spécifique de ces comorbidités doit donc être envisagée à chaque étape du parcours de soins.
Les récidives fréquentes justifient un suivi à vie
Le suivi au long cours de ces patients est indispensable pour dépister d’éventuelles récidives. Dans le cas particulier de la maladie de Cushing, une récidive survient dans 10 à 20 % des cas, parfois plusieurs années après la rémission. Une surveillance au minimum annuelle est donc recommandée les dix premières années après guérison. Elle repose sur l’examen clinique des patients et les tests de dépistage de première intention. Le dosage du cortisol salivaire vespéral, lorsqu’il est disponible, ou le test de freinage minute à la dexaméthasone sont à privilégier au cours de la surveillance, car ils sont les premiers à se modifier en cas de récidive.
Que dire à vos patients ?
Le syndrome de Cushing est une maladie rare dont le diagnostic et le traitement doivent être réalisés en centre expert.
Le traitement du syndrome de Cushing passe par le contrôle de l’excès de cortisol et la prise en charge des comorbidités induites par l’hypercortisolisme (diabète, hypertension artérielle, amyotrophie, troubles psychiatriques...).
Le syndrome de Cushing nécessite un suivi à vie.
Il est exceptionnellement de cause génétique.
Le succès chirurgical ou les traitements anticortisoliques exposent au risque d’insuffisance corticotrope, nécessitant une prise en charge et une éducation spécifique.
Après guérison, les séquelles liées à l’exposition chronique au cortisol doivent être recherchées et prises en charge en centre spécialisé.
Pour en savoir plus
Plusieurs ressources sont à la disposition des patients et des professionnels :
- Centre de référence des maladies rares de la surrénale (CRMRS), CHU de Bordeaux (https ://bit.ly/4q43sJn)
- CRMR de la surrénale, hôpital Cochin (AP-HP) [https ://maladiesrares-cochin-hotel-dieu.aphp.fr/surrenale/]
- CRMR de l’hypophyse, hôpital de la Conception (AP-HM) [https ://fr.ap-hm.fr/site/defhy]
- Centre de référence des cancers rares de la surrénale, Gustave-Roussy (https ://www.gustaveroussy.fr)
- Réunion de concertation pluridisciplinaire (RCP) de recours : RCP nationale FIRENDO pathologies surrénaliennes bénignes et rares ; RCP nationale HYPO
- Protocole national de diagnostic et de soins du syndrome de Cushing (https ://bit.ly/3MvsXEW)
- Société française d’endocrinologie (https ://www.sfendocrino.org)
- Filière maladies rares endocriniennes (https ://www.firendo.fr)
- Surrénales, association de patients (https ://www.surrenales.com/)
2. Pivonello R, Isidori AM, de Martino MC, et al. Complications of Cushing’s syndrome: State of the art. Lancet Diabetes Endocrinol 2016;4(7):611-29.
3. Centre de référence des maladies rares de la surrénale. Protocole national de diagnostic et de soins (PNDS). Syndrome de Cushing. 2022. https://bit.ly/3Yh68HI
4. Tabarin A, Assie G, Barat P, et al. Consensus statement by the French Society of Endocrinology (SFE) and French Society of Pediatric Endocrinology & Diabetology (SFEDP) on diagnosis of Cushing’s syndrome. Ann Endocrinol (Paris) 2022;83(2):119-41.
5. Ferriere A, Tabarin A. Current challenges in Cushing’s syndrome testing: Blood, saliva, urine, or hair? Curr Opin Endocrinol Diabetes Obes 2025;32(5):233-9.
6. Young J, Haissaguerre M, Viera-Pinto O, et al. Management of endocrine disease: Cushing’s syndrome due to ectopic ACTH secretion: An expert operational opinion. Eur J Endocrinol 2020;182(4):R29-58.
7. Lavoillotte J, Mohammedi K, Salenave S, et al. Personalized noninvasive diagnostic algorithms based on urinary free cortisol in ACTH-dependant Cushing’s syndrome. J Clin Endocrinol Metab 2024;109(11):2882-91.
8. Nieman LK, Biller BMK, Findling JW, et al. Treatment of Cushing’s syndrome: An endocrine society clinical practice guideline. J Clin Endocrinol Metab 2015;100(8):2807-31.
9. Réseau ENDOCAN-COMETE. Corticosurrénalome – thésaurus (recommandations). 2014. https://bit.ly/4oRqiTF
Dans cet article
- Épidémiologie et formes étiologiques
- Des symptômes le plus souvent non spécifiques
- Modalités diagnostiques et d’exploration
- Dépistage du syndrome de Cushing en ambulatoire
- Diagnostic étiologique
- Prise en charge thérapeutique
- Prise en charge des complications
- Les récidives fréquentes justifient un suivi à vie
 Encadrés
Encadrés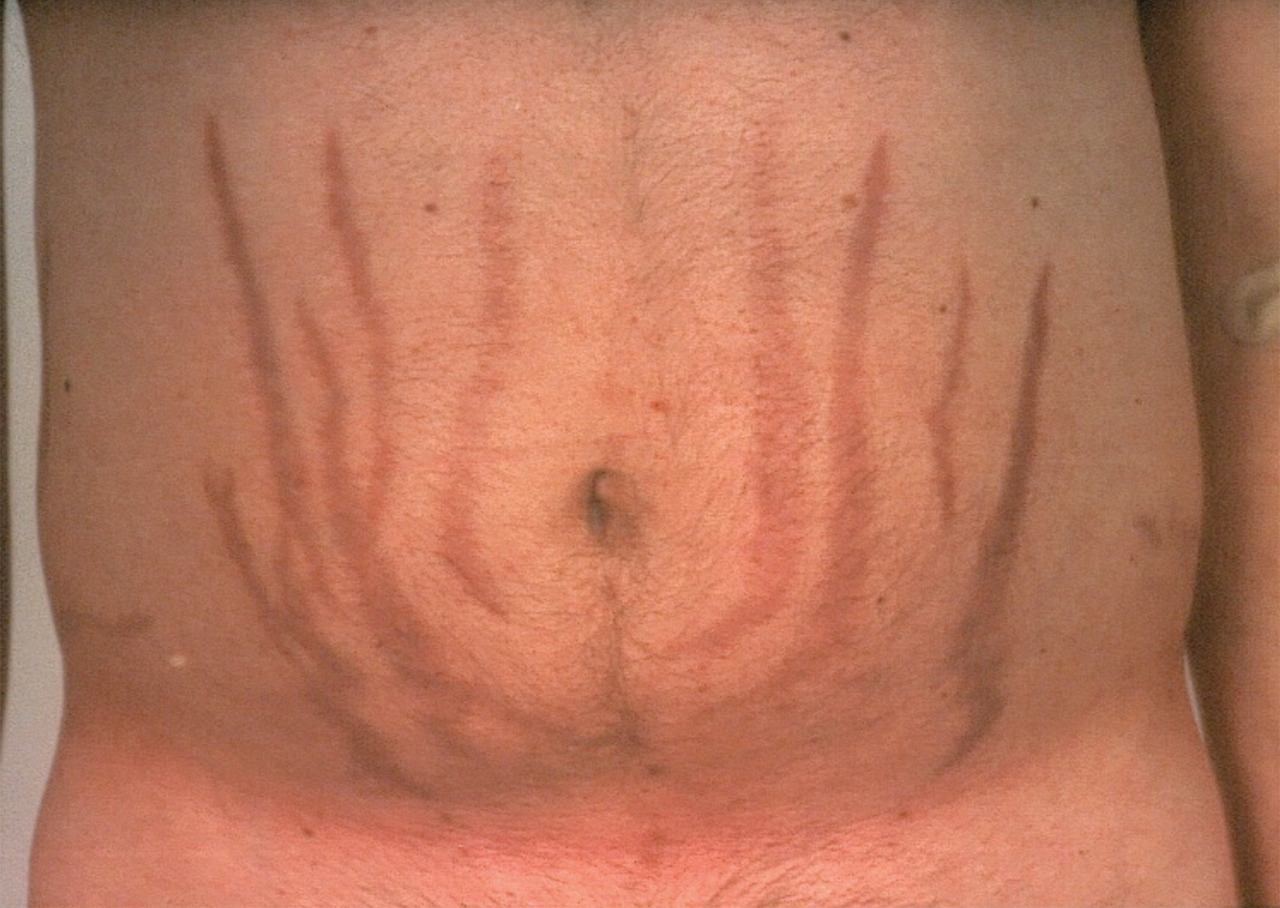



Le syndrome de Cushing est recherché en cas de signes cliniques multiples et/ou progressifs, en particulier ceux dits « spécifiques », chez les patients ayant des comorbidités inhabituelles pour leur âge (ostéoporose, hypertension, diabète chez les sujets jeunes...) et chez ceux ayant un incidentalome surrénalien.
Le dépistage biologique repose sur trois tests : le test de freinage minute à la dexaméthasone, le cortisol libre urinaire des vingt-quatre heures et le cortisol salivaire vespéral.
Le bilan étiologique doit être orienté par le dosage de l’ACTH plasmatique. Les examens d’imagerie sont interprétés en fonction du caractère ACTH-dépendant ou non du syndrome de Cushing.
La chirurgie de la tumeur causale est le traitement de première intention dans la grande majorité des cas.
Le syndrome de Cushing, même guéri, nécessite un suivi à long terme afin d’éliminer une récidive.