À la fin de l’année 2020, une équipe américaine a rapporté pour la première fois un nouveau syndrome auto-inflammatoire acquis : le VEXAS, pour « vacuoles, enzyme E1, lié au chromosome X, auto-inflammatoire, et mutations somatiques ».1 Contrairement à la plupart des syndromes génétiques qui sont généralement identifiés sur la base de phénotypes cliniques homogènes, le VEXAS est unique en ce sens qu’il est issu d’une étude génotypique avec approche « genotype-first ». À partir d’une analyse génétique sur 2 560 patients présentant des fièvres récurrentes, des maladies systémiques inexpliquées ou des symptômes atypiques ou non classés, trois hommes porteurs d’une mutation dans le gène UBA1 (ubiquitin-activating enzyme E1) ont été identifiés. Une recherche de cette même mutation UBA1 chez des patients ayant un phénotype clinique similaire a permis d’identifier un total de 25 patients, définissant ainsi le VEXAS. Initialement considéré comme une maladie rare, le VEXAS est probablement sous-diagnostiqué, avec une prévalence d’environ 1 pour 4 000 chez les hommes de plus de 50 ans et une pénétrance de 100 %.2
Physiopathologie du VEXAS : de la mutation génétique somatique à l’auto-inflammation
La mutation responsable du syndrome VEXAS est le plus souvent une mutation faux-sens du codon 41 de l’exon 3 du gène UBA1, situé sur le chromosome X. Cette mutation est somatique (c’est-à-dire acquise), limitée au compartiment hématopoïétique, présente non seulement dans les précurseurs médullaires des cellules myéloïdes et lymphoïdes mais aussi dans les cellules myéloïdes matures, telles que les monocytes, les neutrophiles, tout en étant absente dans les lymphocytes matures.1 Le gène UBA1 code pour l’enzyme E1 impliqué dans le système ubiquitine-protéasome responsable de la protéolyse intracellulaire, une fonction cruciale facilitant la dégradation des protéines. Dans le syndrome VEXAS, la traduction de la protéine à partir du gène UBA1 muté conduit à une isoforme pathologique inactive d’UBA1, entraînant une accumulation des protéines par une perturbation du mécanisme d’ubiquitination-protéolyse qui induit un stress cellulaire aboutissant finalement à une inflammation incontrôlée.
Manifestations inflammatoires
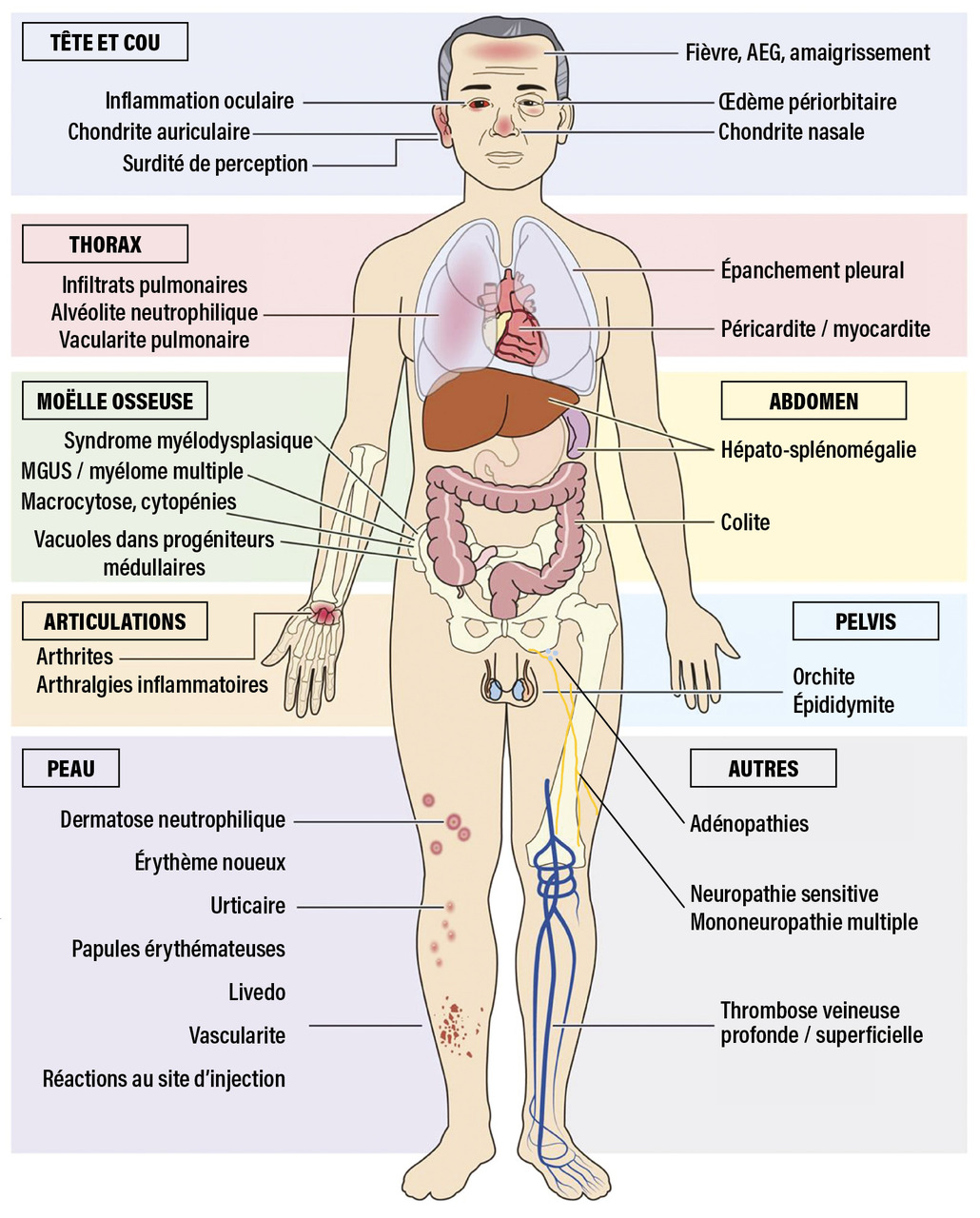
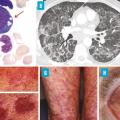
Dans la description initiale, les patients étaient tous des hommes, avec un âge médian au début de la maladie de 64 ans.1 La plupart d’entre eux présentaient des épisodes récurrents de fièvre, une atteinte cutanée, un infiltrat pulmonaire, des chondrites et des thromboses (fig. 1). Un syndrome inflammatoire important était observé, avec des niveaux élevés de protéine C-réactive (CRP), TNFα, IL- 6 et interféron-γ. La plupart des patients répondaient aux critères cliniques d’une autre maladie inflammatoire (polychondrite atrophiante, syndrome de Sweet, périartérite noueuse ou artérite à cellules géantes) ou d’une affection hématologique (syndrome myélodysplasique [SMD], myélome multiple [MM] ou gammapathie monoclonale de signification indéterminée [MGUS]).
Depuis, grâce à plusieurs cohortes dans le monde – la plus importante étant la française, avec 116 patients –, le spectre clinique et biologique s’est étendu et précisé.3 - 5
Le syndrome VEXAS a également été documenté chez environ une dizaine de femmes, la majorité d’entre elles présentant une monosomie acquise du chromosome X ou le syndrome de Turner.11 La présentation clinique est comparable à celle observée chez les hommes.
Les principales caractéristiques cliniques et leur fréquence sont résumées dans le tableau.
Signes cutanés précoces et fréquents
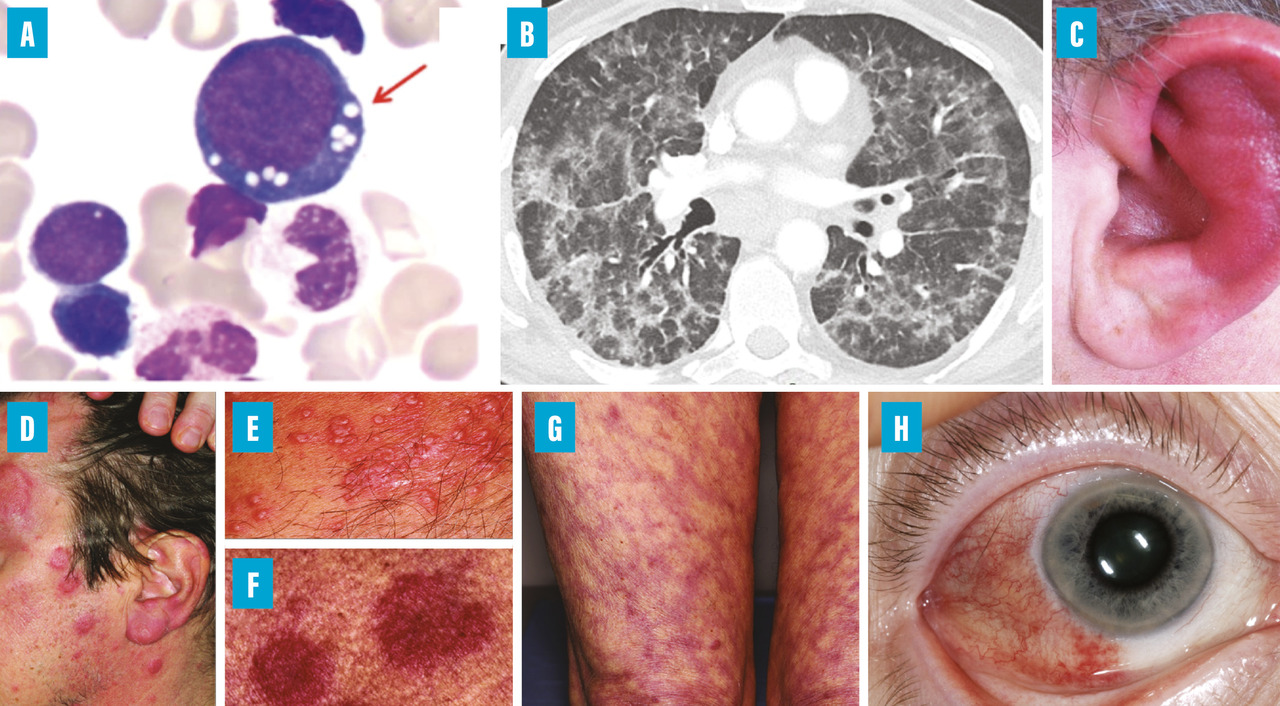
L’atteinte cutanée semble être l’atteinte la plus fréquente et souvent la première manifestation du VEXAS. Dans une série française, les patients présentaient des lésions cutanées de dermatose neutrophilique, comprenant des papules rouges ou violacées douloureuses, parfois œdémateuses, des papules œdémateuses inflammatoires au niveau du cou et du tronc (parfois ombiliquées) et des plaques et nodules érythémateux purpuriques ou pigmentés et infiltrés fermes ; certains présentaient également un livedo racemosa (fig. 2).6 Les analyses histologiques ont identifié des infiltrats périvasculaires composés de lymphohistiocytes périvasculaires, de neutrophiles matures avec leucocytoclasie (mais sans véritables signes de vascularite), associés à des cellules myéloïdes immatures et des signes de dysplasie.
Atteintes pulmonaires multiformes
Les manifestations pulmonaires sont fréquentes au cours du VEXAS, même si elles ne sont généralement pas le problème clinique principal. Une étude française a documenté une atteinte pleuropulmonaire chez 45 patients atteints du VEXAS, avec 44 % de dyspnée, 40 % de toux, mais seulement 7 % nécessitant de l’oxygène.7 Tous les patients présentaient des opacités pulmonaires au scanner thoracique, comprenant des opacités en verre dépoli (87 %), des consolidations (49 %), des réticulations (38 %) et des lignes septales (51 %). Un épanchement pleural était observé chez 53 % des patients et des adénomégalies médiastinales chez 58 %. Les lésions ne semblaient pas évoluer vers une fibrose pulmonaire.
Manifestations thrombotiques, plutôt veineuses
Des événements thrombotiques, majoritairement veineux, sont observés chez 35 à 45 % des patients, incluant des thromboses veineuses profondes et superficielles, des thromboses parfois récurrentes, et ce malgré une anticoagulation.8
Les thromboses artérielles sont possibles mais rares (1,6 %).
Chondrites du nez et du pavillon de l’oreille pour un patient sur trois
Des chondrites du nez et du pavillon de l’oreille sont observées chez un tiers des patients. Contrairement aux polychondrites atrophiantes (PCA) idiopathiques, les chondrites des voies aériennes ou chondrocostales sont rares ou absentes. Chez un patient avec un phénotype de PCA, la présence simultanée des trois critères (sexe masculin, volume globulaire moyen [VGM] supérieur à 100 fL et numération plaquettaire inférieure à 200 G/L) permet de prédire un diagnostic de VEXAS avec une sensibilité de 100 % et une spécificité de 96 %.9
Atteintes oculaires caractéristiques
L’atteinte oculaire est également fréquente au cours du VEXAS (30 à 57 %).3 - 5 Les principales manifestations comprennent l’épisclérite, la sclérite et l’uvéite, tandis que l’inflammation orbitaire et périorbitaire est moins fréquente. Néanmoins, l’œdème périorbitaire reste la présentation la plus caractéristique.
Infections graves fréquemment associées
Des infections graves sont enfin fréquemment associées au syndrome VEXAS. Dans une étude rétrospective française portant sur 74 patients atteints de 133 infections graves, les sites d’infection les plus courants étaient les poumons (59 %), la peau (10 %) et les voies urinaires (9 %). Une confirmation microbiologique a été obtenue dans 76 % des cas : 52 % bactérienne, 30 % virale, 15 % fongique et 3 % mycobactérienne. La forte incidence d’infections atypiques telles que la légionellose et les infections fongiques invasives, survenant malgré une prophylaxie anti-infectieuse ou chez des patients sans traitement immunosuppresseur, peut suggérer un déficit immunitaire intrinsèque à la maladie.10
Manifestations hématologiques
Une anémie macrocytaire d’origine plurifactorielle (inflammation, dysérythropoïèse, iatrogène) est retrouvée dans plus de 90 % des cas, avec parfois une dépendance transfusionnelle.
La thrombopénie est variable selon les séries, rarement hémorragique.
Une neutropénie est rare (moins de 30 % des cas), tandis que la lymphopénie et la monocytopénie sont fréquentes.
La présence de vacuoles dans les progéniteurs myéloïdes et érythroïdes au myélogramme est un élément caractéristique mais non spécifique du syndrome VEXAS (fig. 2A).12
Un syndrome myélodysplasique (SMD) est fréquemment observé dans le syndrome VEXAS : 25 à 55 % en fonction des séries.12 Contrairement aux SMD classiques, le SMD du VEXAS est caractérisé par une dysplasie modérée avec peu ou pas d’excès de blastes, un caryotype normal dans la majorité des cas, un score pronostique le plus souvent favorable selon les classifications habituelles et l’absence de transformation en leucémie aiguë myéloïde. Cependant, des mutations additionnelles classiques d’hématopoïèse clonale comme DNMT3A et/ou TET2, fréquemment associées aux hémopathies myéloïdes, sont fréquentes (60 % des cas) et associées à un risque accru de mortalité.13
Enfin, des MGUS sont observées dans 10 à 20 % des situations et quelques cas de myélomes ont été rapportés.12
Diagnostic génétique
La confirmation diagnostique du syndrome VEXAS est génétique et repose sur l’identification d’une mutation dans le gène UBA1 à partir d’échantillons biologiques (sang ou moelle osseuse). Ce test génétique doit être envisagé chez les hommes de plus de 50 ans présentant un phénotype clinique compatible, associé à une macrocytose.
Plusieurs techniques peuvent être utilisées. Le séquençage de type Sanger ciblant les mutations les plus fréquentes est une méthode rapide et peu coûteuse. L’analyse par séquençage NGS (next-generation sequencing) de panels de gènes est plus sensible mais plus longue et coûteuse ; elle permet en outre l’analyse simultanée d’autres gènes myéloïdes.
Prise en charge thérapeutique empirique
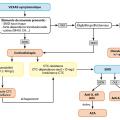
Le pronostic du syndrome VEXAS est sévère, avec une survie de 60 % à cinq ans, notamment du fait des infections.3 En l’absence d’essais prospectifs publiés, la prise en charge thérapeutique des patients avec un syndrome VEXAS est actuellement mal codifiée et s’appuie sur les données de petites études rétrospectives et l’avis d’experts. Le traitement repose sur deux approches principales : cibler les voies inflammatoires ou la population clonale hématopoïétique mutée UBA1.
Traitements visant l’inflammation
Les corticoïdes sont généralement utilisés en première ligne. Les manifestations inflammatoires sont typiquement sensibles aux corticoïdes à fortes doses, mais souvent au prix d’un niveau élevé de dépendance nécessitant des agents épargneurs en deuxième ligne.
Les immunosuppresseurs conventionnels tels que le méthotrexate, l’azathioprine ou le cyclophosphamide ont été utilisés avec peu ou pas d’efficacité.
Les biothérapies ciblant les cytokines pro-inflammatoires peuvent être bénéfiques pour les patients aux manifestations principalement inflammatoires.14
L’anakinra, un inhibiteur du récepteur de l’interleukine (IL) - 1, a été utilisé chez quelques patients avec des résultats mitigés, et surtout des interruptions fréquentes en raison de réactions cutanées au site d’injection.
Le tocilizumab, un inhibiteur du récepteur de l’IL- 6, a montré son efficacité dans plusieurs séries permettant dans certains cas d’arrêter la corticothérapie avec une tolérance plutôt satisfaisante.
Une étude rétrospective internationale portant sur 30 patients a évalué l’efficacité des inhibiteurs de Janus kinases (JAKi) et a rapporté des taux de réponse clinique de 50 % à un mois et d’environ 30 % à six mois, le ruxolitinib montrant une réponse supérieure à six mois par rapport aux autres JAKi.15 Cependant, les effets indésirables des JAKi doivent être pris en compte, en particulier les infections graves et les événements thrombotiques dans cette population âgée avec des comorbidités.
D’autres travaux rétrospectifs et prospectifs sont en cours pour comparer ces différentes stratégies ciblant l’inflammation.
Ciblage de la population clonale
De façon similaire aux traitements des SMD classiques, les agents hypométhylants tels que l’azacitidine (AZA) ont été utilisés pour traiter les patients VEXAS avec SMD concomitant. Dans une série rétrospective de 11 patients, l’AZA a entraîné une réponse clinique chez 5 patients (46 %) et permis une diminution significative des corticoïdes avec des durées de réponse allant de six à plus de vingt-sept mois.16 Dans un essai prospectif chez 30 patients atteints de manifestations inflammatoires associées aux SMD (incluant 12 patients VEXAS), l’AZA permettait d’obtenir une réponse inflammatoire chez 66 % des patients et une réponse hématologique chez 59 %.17
L’allogreffe de cellules souches hématopoïétiques est actuellement le seul traitement curatif du VEXAS ; toutefois, seules des petites séries ont été publiées. Dans la seule étude prospective, cinq patients allogreffés étaient en vie et en rémission après un suivi médian de 9,6 mois sans réaction sévère du greffon contre l’hôte.18
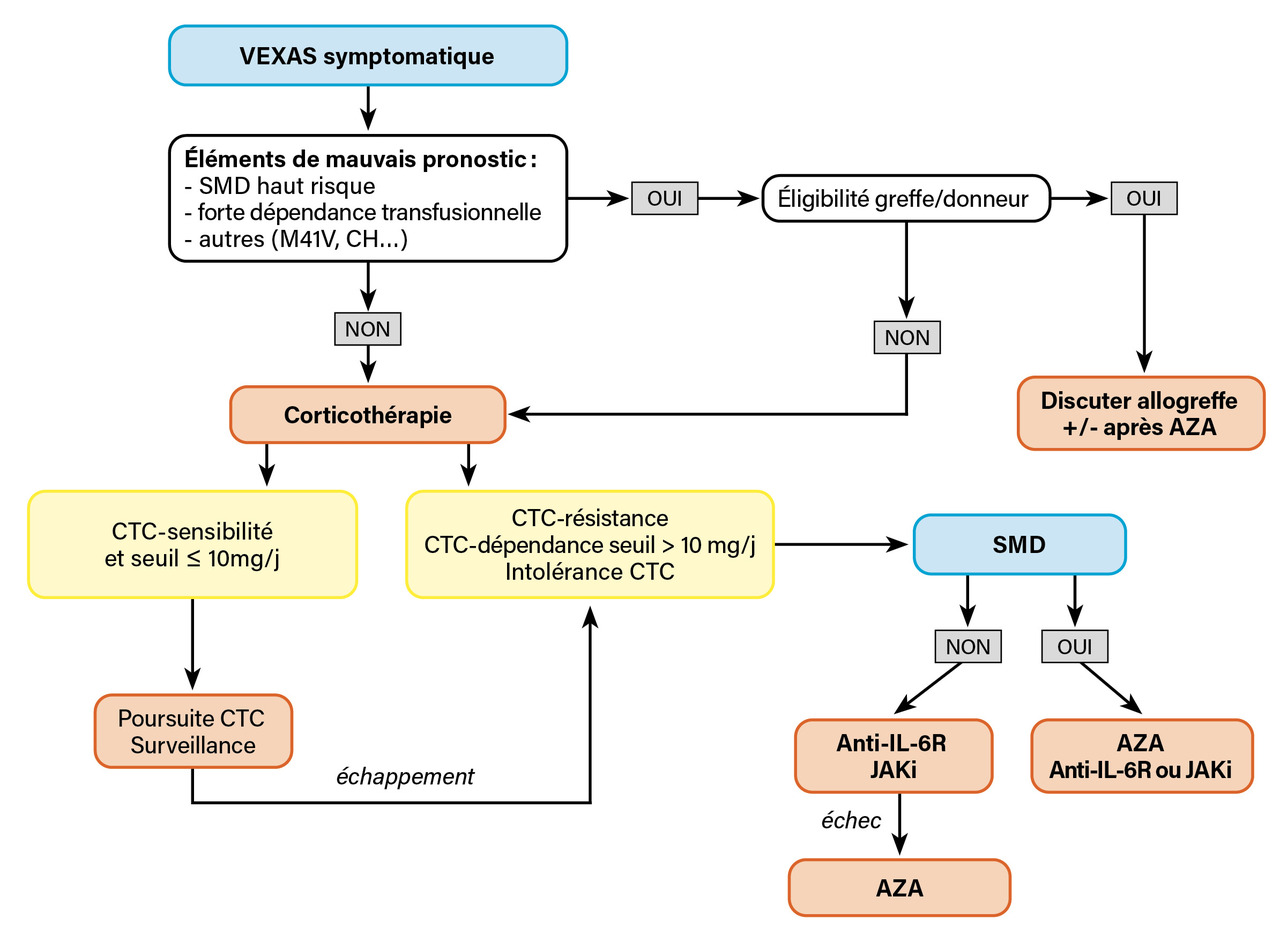
Une proposition non validée de prise en charge thérapeutique fondée sur l’avis d’experts est proposée dans la figure 3.
Les soins de support sont également importants et comprennent les transfusions de globules rouges ou plaquettes, les agents stimulant l’érythropoïèse, la prophylaxie antithrombotique et la prophylaxie anti-infectieuse, bien qu’aucune étude n’ait évalué l’efficacité de ces mesures.
Syndrome en cours d’élucidation
Le VEXAS est un syndrome auto-inflammatoire causé par une mutation somatique dans le gène UBA1. Depuis la description initiale, des progrès significatifs ont été réalisés dans la compréhension de ses mécanismes physiopathologiques, son épidémiologie, son phénotype clinique et l’identification de thérapeutiques prometteuses. Des recommandations sur le diagnostic et la prise en charge du syndrome VEXAS sont en cours d’élaboration par un groupe international d’experts. Elles contribueront notamment à définir les objectifs thérapeutiques et à proposer des critères de réponse pour les différentes composantes de la maladie (inflammatoire, hématologique et moléculaire) afin de faciliter la mise en œuvre d’essais cliniques à grande échelle.
2. Beck DB, Bodian DL, Shah V, et al. Estimated prevalence and clinical manifestations of UBA1 variants associated with VEXAS syndrome in a clinical population. JAMA 2023;329(4):318‑24.
3. Georgin-Lavialle S, Terrier B, Guedon AF, et al. Further characterization of clinical and laboratory features in VEXAS syndrome: Large-scale analysis of a multicentre case series of 116 French patients. Br J Dermatol 2022;186(3):564-74.
4. Ferrada MA, Savic S, Cardona DO, et al. Translation of cytoplasmic UBA1 contributes to VEXAS syndrome pathogenesis. Blood 2022;140(13):1496‑506.
5. Maeda A, Tsuchida N, Uchiyama Y, et al. Efficient detection of somatic UBA1 variants and clinical scoring system predicting patients with variants in VEXAS syndrome. Rheumatology (Oxford) 2024;63(8):2056-64.
6. Zakine È, Papageorgiou L, Bourguiba R, et al. Clinical and pathological features of cutaneous manifestations in VEXAS syndrome: A multicenter retrospective study of 59 cases. J Am Acad Dermatol 2023;88(4):917-20.
7. Borie R, Debray MP, Guedon AF, et al. Pleuropulmonary manifestations of Vacuoles, E1 Enzyme, X-Linked, Autoinflammatory, Somatic (VEXAS) syndrome. Chest 2023;163(3):575‑85.
8. Oo TM, Koay JTJ, Lee SF, et al. Thrombosis in VEXAS syndrome. J Thromb Thrombolysis 2022;53(4):965‑70.
9. Ferrada MA, Sikora KA, Luo Y, et al. Somatic mutations in UBA1 define a distinct subset of relapsing polychondritis patients with VEXAS. Arthritis Rheumatol 2021;73(10):1886‑95.
10. de Valence B, Delaune M, Nguyen Y, et al. Serious infections in patients with VEXAS syndrome: Data from the French VEXAS registry. Ann Rheum Dis 2024;83(3):372-81.
11. Arlet JB, Terrier B, Kosmider O. Mutant UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med 2021;384(22):2163.
12. Koster MJ, Lasho TL, Olteanu H, et al. VEXAS syndrome: Clinical, hematologic features and a practical approach to diagnosis and management. Am J Hematol 2024;99(2):284-99.
13. Gutierrez-Rodrigues F, Kusne Y, Fernandez J, et al. Spectrum of clonal hematopoiesis in VEXAS syndrome. Blood 2023;142(3):244‑59.
14. Boyadzhieva Z, Ruffer N, Kötter I, et al. How to treat VEXAS syndrome: A systematic review on effectiveness and safety of current treatment strategies. Rheumatology (Oxford) 2023;62(11):3518‑25.
15. Heiblig M, Ferrada MA, Koster MJ, et al. Ruxolitinib is more effective than other JAK inhibitors to treat VEXAS syndrome: A retrospective multicenter study. Blood 2022;140(8):927‑31.
16. Comont T, Heiblig M, Riviere E, et al. Azacitidine for patients with Vacuoles, E1 Enzyme, X-linked, Autoinflammatory, Somatic syndrome (VEXAS) and myelodysplastic syndrome: Data from the French VEXAS registry. Br J Haematol 2022;196(4):969‑74.
17. Mekinian A, Zhao LP, Chevret S, et al. A phase II prospective trial of azacitidine in steroid-dependent or refractory systemic autoimmune/inflammatory disorders and VEXAS syndrome associated with MDS and CMML. Leukemia 2022;36(11):2739‑42.
18. Mangaonkar AA, Langer KJ, Lasho TL, et al. Reduced intensity conditioning allogeneic hematopoietic stem cell transplantation in VEXAS syndrome: Data from a prospective series of patients. Am J Hematol 2023;98(2):E28‑31.