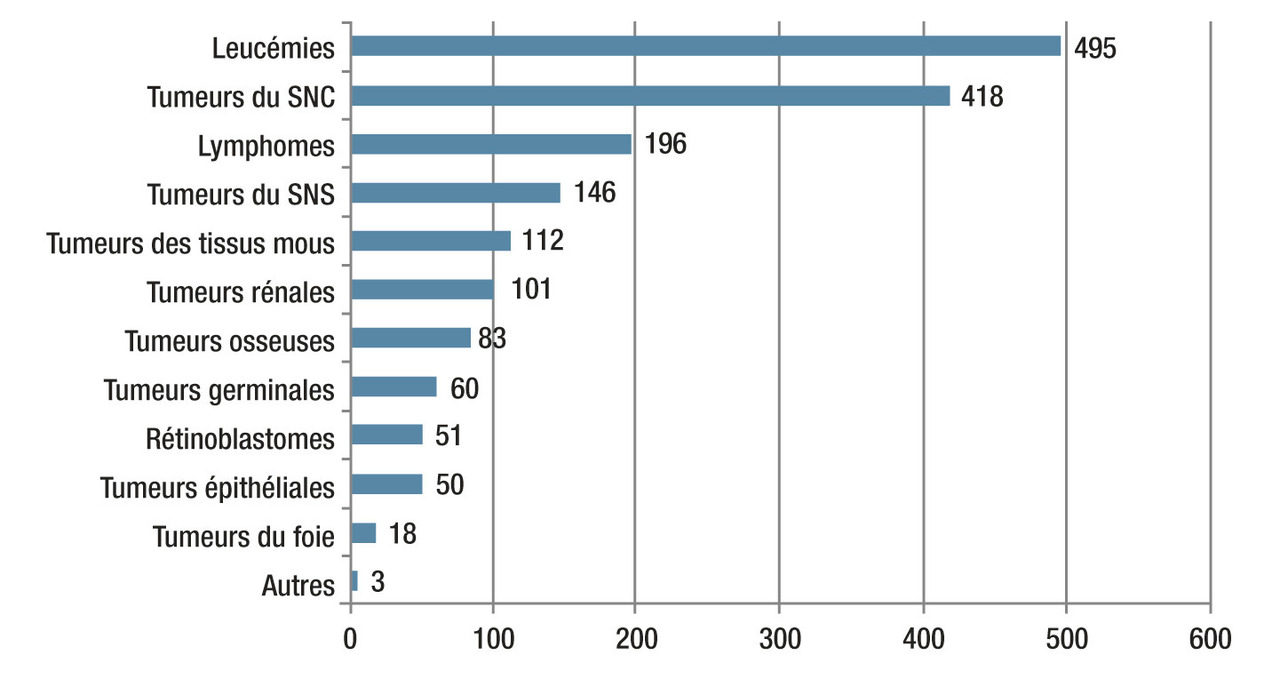
Les leucémies représentent un tiers des cancers pédiatriques avec environ 500 nouveaux cas par an en France.1 Pour la très grande majorité, aucun facteur favorisant génétique ou environnemental n’est mis en évidence, la maladie survenant chez un enfant jusqu’alors sans antécédents particuliers. Dans une minorité de cas, des déterminants génétiques sont identifiés. Le rôle de causes environnementales dans l’émergence des leucémies n’est pas clairement démontré. Seules les expositions aux radiations ionisantes (radiothérapies, accidents nucléaires) et à certaines chimiothérapies cytotoxiques (étoposide, anthracyclines, alkylants) sont responsables d’une augmentation de l’incidence chez les sujets concernés.
De quoi parle-t-on ?
On appelle leucémies différents types d’hémopathies malignes qui ont pour origine la transformation maligne d’une cellule hématopoïétique, responsable d’un envahissement de la moelle osseuse.
Les leucémies aiguës (LA) sont liées à l’expansion clonale de précurseurs médullaires qui, bloqués à un stade précoce de leur différenciation, ne peuvent plus devenir des cellules sanguines matures. L’amplification de ce clone se fait aux dépens de la moelle normale aboutissant à une insuffisance médullaire. Une LA correspond à un envahissement par des blastes supérieur ou égal à 20 %. Elles représentent la quasi- totalité des leucémies de l’enfant. Elles sont lymphoblastiques (LAL) dans 80 % des cas et myéloblastiques (LAM) dans 20 %. Les LAL ont un meilleur pronostic que les LAM. Actuellement, la survie à 5 ans est estimée à respectivement 90 % et 65 %.2, 3
Parmi les LAL, celles de la lignée B prédominent (85 %) avec un pic de fréquence marqué entre 2 et 5 ans. Celles de la lignée T sont rares avant 5 ans et concernent surtout les préadolescents et adolescents de sexe masculin. Les LAM touchent toutes les tranches d’âge sans prédominance.
Les leucémies chroniques de l’enfant sont la leucémie myéloïde chronique4 et la leucémie myélomonocytaire juvénile5 (soit moins de 5 % des leucémies de l’enfant). Elles requièrent une prise en charge tout à fait spécifique.
Les leucémies aiguës (LA) sont liées à l’expansion clonale de précurseurs médullaires qui, bloqués à un stade précoce de leur différenciation, ne peuvent plus devenir des cellules sanguines matures. L’amplification de ce clone se fait aux dépens de la moelle normale aboutissant à une insuffisance médullaire. Une LA correspond à un envahissement par des blastes supérieur ou égal à 20 %. Elles représentent la quasi- totalité des leucémies de l’enfant. Elles sont lymphoblastiques (LAL) dans 80 % des cas et myéloblastiques (LAM) dans 20 %. Les LAL ont un meilleur pronostic que les LAM. Actuellement, la survie à 5 ans est estimée à respectivement 90 % et 65 %.2, 3
Parmi les LAL, celles de la lignée B prédominent (85 %) avec un pic de fréquence marqué entre 2 et 5 ans. Celles de la lignée T sont rares avant 5 ans et concernent surtout les préadolescents et adolescents de sexe masculin. Les LAM touchent toutes les tranches d’âge sans prédominance.
Les leucémies chroniques de l’enfant sont la leucémie myéloïde chronique4 et la leucémie myélomonocytaire juvénile5 (soit moins de 5 % des leucémies de l’enfant). Elles requièrent une prise en charge tout à fait spécifique.
Organisation des soins
La prise en charge des leucémies aiguës repose sur des équipes spécialisées, organisées pour assurer des soins continus au sein d’un réseau bien coordonné.
En France, 31 centres spécialisés de soins en cancérologie de l’enfant sont identifiés (sites de radiothérapie oncopédiatrique non comptés) et chacun est inclus au sein d’une des 7 organisations hospitalières interrégionales de recours en oncologie pédiatrique validées par l’Institut national du cancer. Ces centres sont bien répartis sur l’ensemble du territoire à l’exception de l’outre-mer où seule la Réunion en possède un.
Sitôt le diagnostic établi, le patient est accueilli au sein d’une des structures spécialisées, qui organise les soins avec le service de pédiatrie du centre hospitalier à proximité du domicile du patient, et également avec les professionnels médicaux et non médicaux intervenant au domicile.
Le traitement spécifique de la LA comporte une combinaison de chimiothérapies cytotoxiques pourvoyeuses d’effets secondaires et de complications, qui requièrent des soins de support (gestion des neutropénies fébriles, douleur aiguë liée à une mucite, nausées et vomissements, besoins transfusionnels, support nutritionnel, etc.). Outre la toxicité aiguë, ces médicaments sont source de séquelles tardives.
La pose d’un cathéter veineux central permet l’administration intraveineuse des traitements et le contrôle répété du bilan sanguin. Lors des phases thérapeutiques les plus intensives, un isolement protecteur est nécessaire en hospitalisation.
Les soins comprennent aussi la prise en charge psychosociale et motrice : kinésithérapie, psychomotricité. La poursuite du parcours scolaire est également organisée, adaptée à l’état du jeune malade, avec dès que possible un retour dans l’établissement d’origine accompagné d’un projet d’accueil individualisé.
En France, 31 centres spécialisés de soins en cancérologie de l’enfant sont identifiés (sites de radiothérapie oncopédiatrique non comptés) et chacun est inclus au sein d’une des 7 organisations hospitalières interrégionales de recours en oncologie pédiatrique validées par l’Institut national du cancer. Ces centres sont bien répartis sur l’ensemble du territoire à l’exception de l’outre-mer où seule la Réunion en possède un.
Sitôt le diagnostic établi, le patient est accueilli au sein d’une des structures spécialisées, qui organise les soins avec le service de pédiatrie du centre hospitalier à proximité du domicile du patient, et également avec les professionnels médicaux et non médicaux intervenant au domicile.
Le traitement spécifique de la LA comporte une combinaison de chimiothérapies cytotoxiques pourvoyeuses d’effets secondaires et de complications, qui requièrent des soins de support (gestion des neutropénies fébriles, douleur aiguë liée à une mucite, nausées et vomissements, besoins transfusionnels, support nutritionnel, etc.). Outre la toxicité aiguë, ces médicaments sont source de séquelles tardives.
La pose d’un cathéter veineux central permet l’administration intraveineuse des traitements et le contrôle répété du bilan sanguin. Lors des phases thérapeutiques les plus intensives, un isolement protecteur est nécessaire en hospitalisation.
Les soins comprennent aussi la prise en charge psychosociale et motrice : kinésithérapie, psychomotricité. La poursuite du parcours scolaire est également organisée, adaptée à l’état du jeune malade, avec dès que possible un retour dans l’établissement d’origine accompagné d’un projet d’accueil individualisé.
Stratification thérapeutique
Les leucémies aiguës sont des maladies hétérogènes dont le traitement est adapté à chaque sous-type et aux critères pronostiques. La durée et la nature des différentes phases thérapeutiques dépendent de l’identification initiale précise du type de la leucémie (obtenue par immunophénotypage, cytogénétique, génétique moléculaire) et les indicateurs d’évolution de la maladie (réponse au traitement). La combinaison de l’ensemble de ces critères conduit à choisir un protocole spécifique à chaque patient, adapté au pronostic. Ces protocoles nationaux ou internationaux sont souvent des études randomisées (tirage au sort entre 2 traitements) visant à répondre aux questions en suspens et à améliorer les résultats.
L’étude de la maladie résiduelle en cytométrie de flux et/ou en génétique moléculaire vise à évaluer l’efficacité du traitement à un stade inframicroscopique (après obtention de la rémission complète* en fin d’induction). Ces techniques peuvent détecter une cellule leucémique sur 10 000 cellules normales. Les protocoles récents tiennent compte de cette réponse au traitement initial pour décider de l’intensité de la thérapie ultérieure.
L’étude de la maladie résiduelle en cytométrie de flux et/ou en génétique moléculaire vise à évaluer l’efficacité du traitement à un stade inframicroscopique (après obtention de la rémission complète* en fin d’induction). Ces techniques peuvent détecter une cellule leucémique sur 10 000 cellules normales. Les protocoles récents tiennent compte de cette réponse au traitement initial pour décider de l’intensité de la thérapie ultérieure.
Prise en charge des LAL
Depuis 2016, le protocole français de prise en charge des LAL des lignées B et T de l’enfant est le CAALL-F01. Les patients sont inclus dans des groupes de traitement distincts, définis en fonction de leurs caractéristiques initiales (âge, nombre de globules blancs, cytogénétique, génétique moléculaire) et de la réponse au traitement, en particulier évaluée par l’étude de la maladie résiduelle en fin d’induction et de consolidation.2 Ce protocole évalue les modalités d’administration d’Oncaspar, forme pégylée (c’est-à-dire retard) de la L-asparaginase, développée pour réduire sa fréquence d’administration et son immunogénicité, en sus des chimiothérapies habituelles et de la corticothérapie.
Différentes phases sont nécessaires :
– l’induction durant environ 5 semaines, destinée à obtenir une rémission complète (RC). Durant cette phase, l’enfant est hospitalisé afin qu’on puisse gérer les complications de la maladie et le traitement initial, qui combine vincristine ± daunorubicine avec une corticothérapie et l’ONCASPAR. Une RC est obtenue dans plus de 95 % des cas. Elle n’est pas synonyme de guérison, dont on ne peut parler qu’à l’issue de l’ensemble du traitement et en l’absence de rechute à 5 ans.
– Les 5 à 10 mois suivants comportent la consolidation, 1 à 2 intensifications pour tous les sujets traités et 1 à 2 séquences de méthotrexate haute dose (voie IV, phase M).
Durant cette période, l’enfant peut rester la plupart du temps à son domicile. Les chimiothérapies (telles que la vincritine, les anthracyclines, l’aracytine, la cyclophosphamide) sont administrées par voie injectable en hôpital de jour (HDJ), associées à des traitements oraux (6-mercaptopurine, 6-thioguanine, méthotrexate et corticothérapie orale).
– Le traitement d’entretien, qui dure 18 à 24 mois, comporte des injections de vincristine espacées en HDJ, auxquelles s’ajoute un traitement cytotoxique oral (6-mercaptopurine en prise quotidienne et méthotrexate 1 fois par semaine). Cette période permet un retour à plein temps de l’enfant dans son milieu scolaire habituel, et s’accompagne de la repousse des cheveux et du retrait du cathéter central, signes physiques les plus visibles de sa maladie. Il est nécessaire de rester très vigilant quant aux éventuels épisodes fébriles.
Durant tout ce protocole, des chimiothérapies intrathécales sont administrées séquentiellement (tous les 10 jours en induction, puis tous les mois et enfin tous les 2 à 3 mois durant le traitement d’entretien) par ponctions lombaires. But : prévenir l’éventuel passage de cellules leucémiques dans le liquide céphalorachidien (où elles se retrouveraient alors protégées des chimiothérapies par la barrière hématoencéphalique) et bien entendu éradiquer un envahissement méningé.
Un accompagnement approprié de la douleur et de l’anxiété généré par cet acte thérapeutique répété est indispensable.
Différentes phases sont nécessaires :
– l’induction durant environ 5 semaines, destinée à obtenir une rémission complète (RC). Durant cette phase, l’enfant est hospitalisé afin qu’on puisse gérer les complications de la maladie et le traitement initial, qui combine vincristine ± daunorubicine avec une corticothérapie et l’ONCASPAR. Une RC est obtenue dans plus de 95 % des cas. Elle n’est pas synonyme de guérison, dont on ne peut parler qu’à l’issue de l’ensemble du traitement et en l’absence de rechute à 5 ans.
– Les 5 à 10 mois suivants comportent la consolidation, 1 à 2 intensifications pour tous les sujets traités et 1 à 2 séquences de méthotrexate haute dose (voie IV, phase M).
Durant cette période, l’enfant peut rester la plupart du temps à son domicile. Les chimiothérapies (telles que la vincritine, les anthracyclines, l’aracytine, la cyclophosphamide) sont administrées par voie injectable en hôpital de jour (HDJ), associées à des traitements oraux (6-mercaptopurine, 6-thioguanine, méthotrexate et corticothérapie orale).
– Le traitement d’entretien, qui dure 18 à 24 mois, comporte des injections de vincristine espacées en HDJ, auxquelles s’ajoute un traitement cytotoxique oral (6-mercaptopurine en prise quotidienne et méthotrexate 1 fois par semaine). Cette période permet un retour à plein temps de l’enfant dans son milieu scolaire habituel, et s’accompagne de la repousse des cheveux et du retrait du cathéter central, signes physiques les plus visibles de sa maladie. Il est nécessaire de rester très vigilant quant aux éventuels épisodes fébriles.
Durant tout ce protocole, des chimiothérapies intrathécales sont administrées séquentiellement (tous les 10 jours en induction, puis tous les mois et enfin tous les 2 à 3 mois durant le traitement d’entretien) par ponctions lombaires. But : prévenir l’éventuel passage de cellules leucémiques dans le liquide céphalorachidien (où elles se retrouveraient alors protégées des chimiothérapies par la barrière hématoencéphalique) et bien entendu éradiquer un envahissement méningé.
Un accompagnement approprié de la douleur et de l’anxiété généré par cet acte thérapeutique répété est indispensable.
Prise en charge des LAM
Comme pour les protocoles de LAL, la stratification du risque dans les LAM pédiatriques est fondée sur les caractéristiques cytogénétiques et de génétique moléculaire, ainsi que sur la réponse au traitement, en particulier évaluée par le suivi de la maladie résiduelle.3
L’approche thérapeutique classique consiste en l’administration de 4 ou 5 cures de chimiothérapies très aplasiantes comme la cytarabine (Aracytine) associée à des anthracyclines. Chacune de ces cures impose une hospitalisation d’environ 1 mois en secteur protégé, la profondeur et la durée de l’aplasie exposant les patients à un risque infectieux, bactérien et fongique important.
Le protocole MyeChild01 est ouvert en France depuis 2018. Parmi les questions auxquelles il vise à répondre : celle de l’apport et des modalités d’administration du gemtuzumab ozogamicine (Mylotarg, anticorps monoclonal anti- CD33 humanisé conjugué à la calichéamicine, agent cytotoxique) durant l’induction en sus des chimiothérapies par cytarabine et anthracyclines.
La RC est obtenue dans 80 % des cas au terme de l’induction. Un traitement d’entretien n’est pas justifié dans ces leucémies.
L’indication d’une greffe de moelle osseuse allogénique est légitimement discutée en première rémission complète dans les LAM ayant des caractéristiques cytogénétiques et moléculaires défavorables (et ce, plus souvent que pour les LAL). C’est la stratégie prévenant le plus efficacement la rechute.
De façon spécifique, les leucémies à promyélocytes LAM M3 (environ 10% des LAM pédiatriques) bénéficient d’un traitement à part du fait de leur sensibilité à l’acide tout-trans rétinoïque (ATRA, Vesanoid) et au trioxyde d’arsenic.
L’approche thérapeutique classique consiste en l’administration de 4 ou 5 cures de chimiothérapies très aplasiantes comme la cytarabine (Aracytine) associée à des anthracyclines. Chacune de ces cures impose une hospitalisation d’environ 1 mois en secteur protégé, la profondeur et la durée de l’aplasie exposant les patients à un risque infectieux, bactérien et fongique important.
Le protocole MyeChild01 est ouvert en France depuis 2018. Parmi les questions auxquelles il vise à répondre : celle de l’apport et des modalités d’administration du gemtuzumab ozogamicine (Mylotarg, anticorps monoclonal anti- CD33 humanisé conjugué à la calichéamicine, agent cytotoxique) durant l’induction en sus des chimiothérapies par cytarabine et anthracyclines.
La RC est obtenue dans 80 % des cas au terme de l’induction. Un traitement d’entretien n’est pas justifié dans ces leucémies.
L’indication d’une greffe de moelle osseuse allogénique est légitimement discutée en première rémission complète dans les LAM ayant des caractéristiques cytogénétiques et moléculaires défavorables (et ce, plus souvent que pour les LAL). C’est la stratégie prévenant le plus efficacement la rechute.
De façon spécifique, les leucémies à promyélocytes LAM M3 (environ 10% des LAM pédiatriques) bénéficient d’un traitement à part du fait de leur sensibilité à l’acide tout-trans rétinoïque (ATRA, Vesanoid) et au trioxyde d’arsenic.
Quand faire une allogreffe de moelle osseuse ?
L’allogreffe de cellules souches hématopoïétiques consiste à remplacer la moelle osseuse malade, siège de la leucémie, par la moelle saine d’un donneur.
L’objectif est notamment d’obtenir une immunothérapie susceptible de contrôler la maladie (le greffon détruisant les cellules leucémiques). Cependant, on doit redouter l’effet greffon contre l’hôte, néfaste. C’est donc une procédure lourde, grevée d’une mortalité liée au traitement de près de 10 %, et responsable de séquelles dont la stérilité due au conditionnement myélo-ablatif précédant la greffe (chimio- et parfois radiothérapie, très toxique à court, moyen et long terme). Elle n’est faisable que chez des patients capables de tolérer l’intensité de la procédure, notamment le traitement immunosuppresseur, et à condition de disposer d’un donneur compatible (idéalement issu de la fratrie). Aussi, un contrôle relatif de la maladie leucémique est nécessaire avant la greffe, en particulier pour les LAL. Toute indication d’allogreffe doit être soigneusement pesée et discutée collégialement.
L’objectif est notamment d’obtenir une immunothérapie susceptible de contrôler la maladie (le greffon détruisant les cellules leucémiques). Cependant, on doit redouter l’effet greffon contre l’hôte, néfaste. C’est donc une procédure lourde, grevée d’une mortalité liée au traitement de près de 10 %, et responsable de séquelles dont la stérilité due au conditionnement myélo-ablatif précédant la greffe (chimio- et parfois radiothérapie, très toxique à court, moyen et long terme). Elle n’est faisable que chez des patients capables de tolérer l’intensité de la procédure, notamment le traitement immunosuppresseur, et à condition de disposer d’un donneur compatible (idéalement issu de la fratrie). Aussi, un contrôle relatif de la maladie leucémique est nécessaire avant la greffe, en particulier pour les LAL. Toute indication d’allogreffe doit être soigneusement pesée et discutée collégialement.
Innovation : où en est-on ?
Dans l’objectif d’améliorer encore le taux de guérison, de nouvelles classes thérapeutiques sont à l’étude. Le principe : contrecarrer la chimiorésistance des cellules leucémiques par des traitements dits « de précision », adaptés à la biologie tumorale. Des agents ciblés visant à atteindre les anomalies moléculaires en cause dans la leucémogenèse sont à l’étude ou déjà utilisés, tels les inhibiteurs de tyrosine kinase (par exemple la midostaurine, Rydapt, dans la LAM avec mutation FLT3).
L’immunothérapie connaît un important essor grâce au développement d’anticorps monoclonaux, en particulier les bispécifiques comme le blinatumomab (Blicynto) anti CD19-CD3 qui active les cellules T endogènes et provoque l’élimination spécifique des cellules B malignes et saines exprimant le CD19.
Également à l’étude : les cellules T modifiées génétiquement (CAR T-cells). Des lymphocytes T issus du patient ou d’un donneur sain sont génétiquement modifiés ex vivo pour exprimer à leur surface un récepteur antigénique chimérique (CAR) ciblant un marqueur présent sur les cellules leucémiques du patient (CD19 par exemple). Elles sont administrées en une injection unique. Ainsi, Kymriah (tisagenlecleucel) a obtenu l’AMM dans la leucémie aiguë lymphoblastique à cellules B.
Les taux de réponses importants observés dans les LAL B CD19+ réfractaires en font une option très prometteuse. à ce jour, un essai est en cours dans un centre pédiatrique français (NCT02808442) et d’autres vont suivre. Les prochaines années, il faudra trouver des solutions aux limites actuelles de ce traitement : accessibilité, coût et gestion de ses effets indésirables propres (syndrome de relargage des cytokines, toxicité neurologique, lymphopénie B persistante).
L’immunothérapie connaît un important essor grâce au développement d’anticorps monoclonaux, en particulier les bispécifiques comme le blinatumomab (Blicynto) anti CD19-CD3 qui active les cellules T endogènes et provoque l’élimination spécifique des cellules B malignes et saines exprimant le CD19.
Également à l’étude : les cellules T modifiées génétiquement (CAR T-cells). Des lymphocytes T issus du patient ou d’un donneur sain sont génétiquement modifiés ex vivo pour exprimer à leur surface un récepteur antigénique chimérique (CAR) ciblant un marqueur présent sur les cellules leucémiques du patient (CD19 par exemple). Elles sont administrées en une injection unique. Ainsi, Kymriah (tisagenlecleucel) a obtenu l’AMM dans la leucémie aiguë lymphoblastique à cellules B.
Les taux de réponses importants observés dans les LAL B CD19+ réfractaires en font une option très prometteuse. à ce jour, un essai est en cours dans un centre pédiatrique français (NCT02808442) et d’autres vont suivre. Les prochaines années, il faudra trouver des solutions aux limites actuelles de ce traitement : accessibilité, coût et gestion de ses effets indésirables propres (syndrome de relargage des cytokines, toxicité neurologique, lymphopénie B persistante).
Références
1. Lacour B, Clavel J. Aspects épidémiologiques des cancers de l’enfant. Rev Prat 2014;64:1264-9.
2. Hunger SP, Mullighan CG. Acute Lymphoblastic Leukemia in Children. N Engl J Med 2015;373:1541-52.
3. Zwaan CM, Kolb EA, Reinhardt D, et al. Collaborative Efforts Driving Progress in Pediatric Acute Myeloid Leukemia. J Clin Oncol 2015;33:2949-62.
4. Millot F, Claviez A, Leverger G, Corbaciglu S, Groll AH, Suttorp M. Imatinib cessation in children and adolescents with chronic myeloid leukemia in chronic phase. Pediatr Blood Cancer 2014;61:355–7.
5. Strullu M, Cavé H. La leucémie myélomonocytaire juvénile. Hématologie 2017;23:122–34.
2. Hunger SP, Mullighan CG. Acute Lymphoblastic Leukemia in Children. N Engl J Med 2015;373:1541-52.
3. Zwaan CM, Kolb EA, Reinhardt D, et al. Collaborative Efforts Driving Progress in Pediatric Acute Myeloid Leukemia. J Clin Oncol 2015;33:2949-62.
4. Millot F, Claviez A, Leverger G, Corbaciglu S, Groll AH, Suttorp M. Imatinib cessation in children and adolescents with chronic myeloid leukemia in chronic phase. Pediatr Blood Cancer 2014;61:355–7.
5. Strullu M, Cavé H. La leucémie myélomonocytaire juvénile. Hématologie 2017;23:122–34.
Dans cet article
essentiel
Cancers pédiatriques les plus fréquents (500 nouveaux cas par an en France).
Taux de guérison : environ 80 %, mais varie selon chaque sous-type. Meilleure survie à 5 ans de la LAL vs la LAM (90 et 65 % respectivement).
Traitement par combinaison de chimiothérapies cytotoxiques adapté à l’identification initiale précise du type et à la maladie résiduelle.
Les anticorps monoclonaux bispécifiques ou les CAR T-cells pour les LAL sont un progrès majeur dont la place doit être précisée.