La drépanocytose est une maladie génétique du globule rouge qui atteint essentiellement les popu- lations d’origine africaine et antillaise. Elle est caractérisée par la production d’une hémoglobine anormale (HbS) entraînant une fragilité des globules rouges, une diminution de leur déformabilité et une augmentation de leur adhérence à l’endothélium vasculaire. Il en résulte une anémie hémolytique chronique, des crises vaso-occlusives pouvant atteindre tous les organes, et une asplénie fonctionnelle. La transfusion est un traitement majeur de la drépanocytose.1 En apportant de l’HbA, elle permet de prévenir et traiter les complications aiguës et chroniques de cette pathologie. Elle n’est cependant pas sans risque chez le patient drépanocytaire. Outre la surcharge en fer, le principal risque de la transfusion au cours de la drépanocytose est l’allo-immunisation anti-érythrocytaire avec pour conséquence un conflit entre globules rouges transfusés et anticorps anti-érythrocytaires du patient, entraînant une hémolyse post-transfusionnelle retardée. Les patients drépanocytaires, polytransfusés, ont un taux d’allo-immunisation élevé par rapport à la population générale, essentiellement du fait du polymorphisme des groupes sanguins entre donneurs, d’origine européenne, en France, et ces patients, d’origine africaine.
Un accident fréquent
L’hémolyse post-transfusionnelle retardée est un accident fréquent chez les patients drépanocytaires, pouvant mettre en jeu le pronostic vital. Elle est sous-diagnostiquée du fait de ses caractéristiques : survenue au décours d’une transfusion (3 à 21 jours), apparition ou récurrence de crises vaso-occlusives, urines foncées et signes d’anémie. Sur le plan biologique, les lactate déshydrogénases augmentent, au-delà des taux habituels observés au cours des complications de la maladie, le taux d’hémoglobine, dans les cas d’hyperhémolyse peut être inférieur au taux prétransfusionnel, une réticulopénie est fréquente, enfin, l’HbA disparaît rapidement, confirmant la destruction des globules rouges transfusés. Le bilan immuno-hématologique peut mettre en évidence des anticorps d’allo-immunisation, témoin du polymorphisme des groupes sanguins entre donneurs et receveurs d’origine différente, mais dans près de 30 % des cas il n’existe pas d’anticorps détectable.2 Il est vivement conseillé de ne pas retransfuser ces patients, sauf si leur pronostic vital est en jeu. En effet, toute nouvelle transfusion peut exacerber le processus hémolytique. Dans les cas sévères, des défaillances multiorganes peuvent aboutir au décès du patient. La prévention et la prise en charge de ces accidents font appel à une collaboration étroite entre les prescripteurs et les professionnels de la transfusion mais aussi à l’éducation thérapeutique des patients, à laquelle doit participer le médecin généraliste.3
Lire aussi | Anémie : quand faut-il transfuser ?🎬
Prévention de l’allo-immunisation et de l’hémolyse post-transfusionnelle
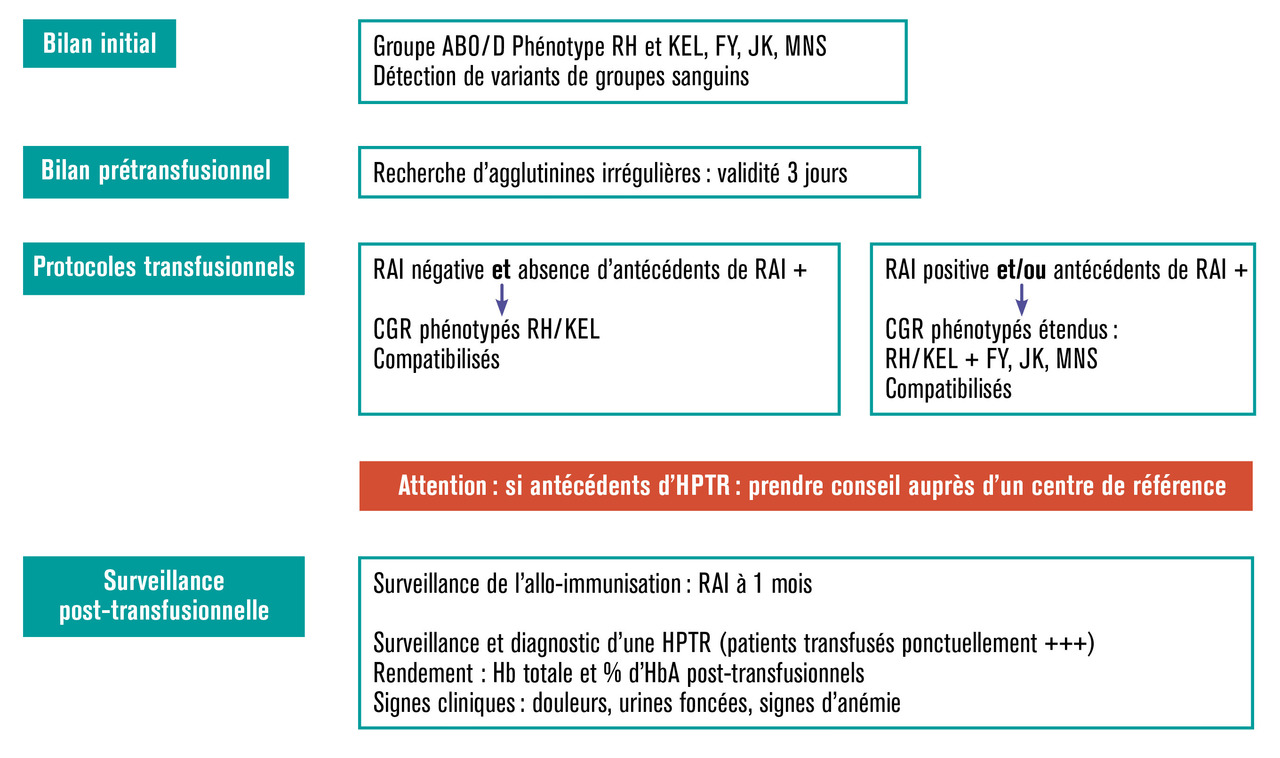
Les protocoles transfusionnels sont adaptés au risque d’allo-immunisation, aux antécédents connus d’hémolyse post-transfusionnelle retardée mais aussi à la ressource disponible en concentrés de globules rouges phénocompatibles. Enfin, le risque de développer ces accidents est beaucoup plus élevé pour des patients transfusés ponctuellement dans un cadre aigu que pour des patients inclus dans un programme transfusionnel chronique.
La connaissance de l’historique transfusionnel des patients et le suivi des immunisations sont fondamentaux. La recherche d’agglutinines irrégulières post-transfusionnelle (1 mois) permet de détecter une immunisation primaire dont il faudra tenir compte pour les transfusions futures afin de prévenir une restimulation et une hémolyse post-transfusionnelle retardée. Un dossier transfusionnel à jour assurera la prise en compte de ces anticorps connus mais non présents le jour de la transfusion, car évanescents. En effet, les anticorps anti-érythrocytaires peuvent apparaître et disparaître, et donc la recherche d’agglutinines irré- gulières du jour ne reflète pas la situation immunologique globale du patient. De plus, les patients ayant déjà eu une hémolyse post-transfusionnelle retardée sont à risque de récurrence de cet accident. La décision transfusionnelle et le choix des concentrés de globules rouges (phénotypé RH/K ou phénotypé étendu aux autres groupes FY, JK, MNS) dépend de ces éléments. Un traitement immunosuppresseur (rituximab) peut être proposé pour les patients allo- immunisés ayant des antécédents d’hémolyse post-transfusionnelle retardée. Cela étant, pour la mise en œuvre de la phénocompatibilité, il est indispensable de disposer de concentrés de globules rouges de donneurs de même origine que les patients, et donc la promotion du don dans ces populations minoritaires est déterminante pour la sécurité.
La connaissance de l’historique transfusionnel des patients et le suivi des immunisations sont fondamentaux. La recherche d’agglutinines irrégulières post-transfusionnelle (1 mois) permet de détecter une immunisation primaire dont il faudra tenir compte pour les transfusions futures afin de prévenir une restimulation et une hémolyse post-transfusionnelle retardée. Un dossier transfusionnel à jour assurera la prise en compte de ces anticorps connus mais non présents le jour de la transfusion, car évanescents. En effet, les anticorps anti-érythrocytaires peuvent apparaître et disparaître, et donc la recherche d’agglutinines irré- gulières du jour ne reflète pas la situation immunologique globale du patient. De plus, les patients ayant déjà eu une hémolyse post-transfusionnelle retardée sont à risque de récurrence de cet accident. La décision transfusionnelle et le choix des concentrés de globules rouges (phénotypé RH/K ou phénotypé étendu aux autres groupes FY, JK, MNS) dépend de ces éléments. Un traitement immunosuppresseur (rituximab) peut être proposé pour les patients allo- immunisés ayant des antécédents d’hémolyse post-transfusionnelle retardée. Cela étant, pour la mise en œuvre de la phénocompatibilité, il est indispensable de disposer de concentrés de globules rouges de donneurs de même origine que les patients, et donc la promotion du don dans ces populations minoritaires est déterminante pour la sécurité.
Lire aussi | Dossier progressif n° 347
Diagnostic d’une hémolyse post-transfusionnelle retardée
La connaissance des signes clinico- biologiques et la démonstration d’une disparition de l’HbA permettent de poser le diagnostic. Il est donc important après toute transfusion (24 à 48 heures), de disposer du taux d’HbA post-transfusionnel pour déterminer à distance si les signes clinico-biologiques observés sont la conséquence d’une hémolyse retardée.
Lire aussi | Dossier progressif n° 314
Traitement d’une hémolyse post-transfusionnelle retardée
Le traitement dépend de la gravité clinico-biologique et doit être pris en charge dans des centres spécialisés pour les formes les plus sévères. L’érythropoïèse doit être suivie de près via le compte des réticulocytes, car les globules rouges drépanocytaires n’ont une durée de vie que de 17 jours, et en l’absence de production la chute du taux d’hémoglobine dans ce contexte peut être très rapide. L’érythropoïèse peut donc être stimulée si besoin par de fortes doses d’érythropoïétine. La retransfusion dépend du seuil de gravité et nécessite d’étendre le phénotype des concentrés de globules rouges, même si aucun anticorps n’est détecté. Certaines molécules ont été utilisées lors d’hémolyse post-transfusionnelle retardée telles les immunoglobulines, les corticoïdes, l’éculizumab. Il n’y a actuellement pas de consensus sur ces traitements, du fait des incertitudes mécanistiques, cependant un faisceau d’évidences sont en faveur de l’activation du complément à la fois via la voie classique et la voie alterne, favorisant l’utilisation de l’éculizumab dans les cas sévères d’hyperhémolyse.
La transfusion des patients drépanocytaires nécessite une prise en charge multidisciplinaire. Une meilleure connaissance des particularités immuno- hématologiques de ces patients et une adéquation des ressources de concentrés de globules rouges phénocom- patibles aux besoins sont des gages d’optimisation de la sécurité transfu- sionnelle (v. figure). Enfin, un effort de recherche est indispensable pour mieux comprendre les mécanismes physiopathologiques de l’allo-immuni- sation et des hémolyses post-trans- fusionnelles retardées au cours de la drépanocytose, permettant une prévention et des traitements spécifiques.
La transfusion des patients drépanocytaires nécessite une prise en charge multidisciplinaire. Une meilleure connaissance des particularités immuno- hématologiques de ces patients et une adéquation des ressources de concentrés de globules rouges phénocom- patibles aux besoins sont des gages d’optimisation de la sécurité transfu- sionnelle (v. figure). Enfin, un effort de recherche est indispensable pour mieux comprendre les mécanismes physiopathologiques de l’allo-immuni- sation et des hémolyses post-trans- fusionnelles retardées au cours de la drépanocytose, permettant une prévention et des traitements spécifiques.
Références
1. Habibi A, Arlet JB, Stankovic K, et al. ; Centre de référence maladies rares. Syndromes drépanocytaires majeurs. Recommandations françaises de prise en charge de la drépanocytose de l’adulte : actualisation 2015. Rev Med Interne 2015;36:5S3-84.
2. Habibi A, Mekontso-Dessap A, Guillaud C, et al. Delayed hemolytic transfusion reaction in adult sickle-cell disease: presentations, outcomes, and treatments of 99 referral center episodes. Am J Hematol 2016;91:989-94.
3. Pirenne F, Yazdanbakhsh K. How I safely transfuse patients with sickle cell disease and manage delayed hemolytic transfusion reactions. Blood 2018;131:2773-81.
2. Habibi A, Mekontso-Dessap A, Guillaud C, et al. Delayed hemolytic transfusion reaction in adult sickle-cell disease: presentations, outcomes, and treatments of 99 referral center episodes. Am J Hematol 2016;91:989-94.
3. Pirenne F, Yazdanbakhsh K. How I safely transfuse patients with sickle cell disease and manage delayed hemolytic transfusion reactions. Blood 2018;131:2773-81.