La nouvelle classe thérapeutique des « caftors » est en train de révolutionner la prise en charge des patients atteints de mucoviscidose. Cette nouvelle thérapie protéique transforme le paradigme de soin dans la mucoviscidose, permet d’envisager une augmentation de l’espérance de vie au-delà de 60 ans et, si cette thérapie est commencée à l’âge pédiatrique, un ralentissement majeur de l’évolution.1
Toutefois, environ 10 % de patients ne peuvent pas en bénéficier.2 Il s’agit principalement de patients porteurs de mutations associées à l’absence de synthèse protéique : soit des mutations non-sens générant un codon stop prématuré (PTC), soit des mutations d’épissage, qui perturbent l’excision des introns dans le noyau et conduisent le plus souvent à des décalages du cadre de lecture et à des PTC. Parfois, même quand la protéine est synthétisée, la protéine mutée n’est pas « corrigée » par ces chaperons pharmacologiques chimiques, et certaines mutations, dites de portail, atteignent la membrane mais présentent une fréquence d’ouverture du canal réduite en raison d’un défaut d’activation et ne répondent pas non plus.3
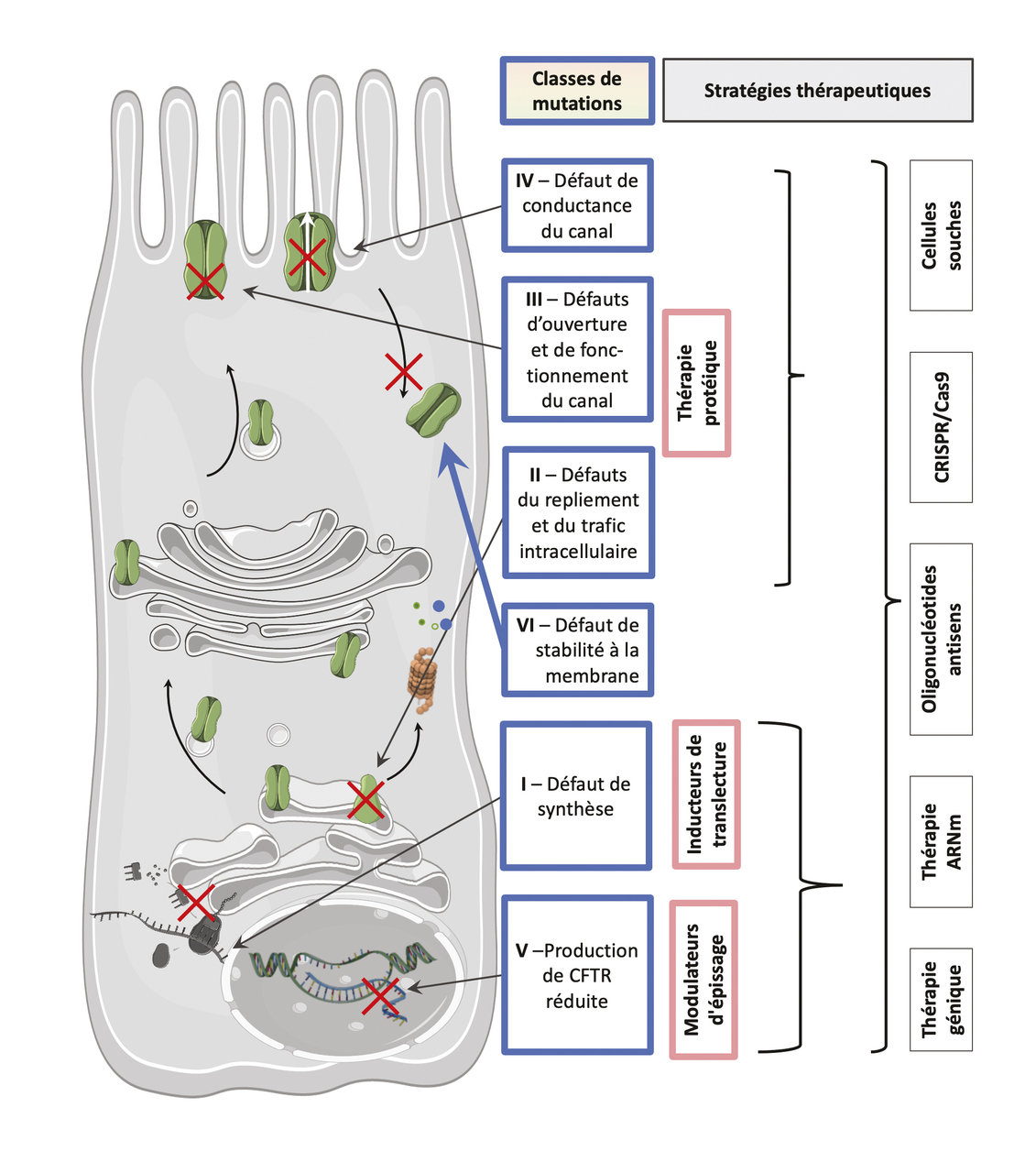
Ainsi, d’autres stratégies sont envisagées. Les thérapies géniques ont pour ambition de corriger le gène CFTR défectueux à l’intérieur même du noyau cellulaire en utilisant des outils moléculaires modernes, tandis que les thérapies de transcrit visent l’ARN messager, par exemple porteur de mutations stop qui induisent un arrêt prématuré de la traduction en protéine (figure).
Thérapie génique pour corriger la mutation
La découverte, en 1989, d’un gène unique, CFTR, à l’origine de la mucoviscidose a ouvert la voie de la thérapie génique.
Les recherches visent l’intégration d’un gène CFTR sain au génome à l’aide de vecteurs viraux ou synthétiques, de type lipidique cationique, polypeptidique soluble ou polymère cationique. Elles présentent de nombreux défis, qui restent encore à surmonter. Le niveau de transfection reste faible, et ce d’autant que les cellules épithéliales respiratoires déjà différenciées sont très difficiles à transfecter. De nouveaux vecteurs plus performants sont à l’étude ; un essai clinique international a débuté avec un pseudotype de lentivirus ayant une enveloppe de virus Sendai favorisant un taux élevé de transfection des cellules respiratoires, une rémanence prolongée, le transport de plus longues séquences d’ADN, et optimisé pour minimiser les réactions d’immunogénicité.4
La réparation spécifique de l’ADN muté par la technologie CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats /CRISPR-associated protein 9) est en plein essor. Le système comprend la nucléase Cas9, qui coupe les deux brins d’ADN à l’endroit spécifié ainsi qu’un ARN guide (ARNg), qui comporte la séquence cible à intégrer.5 D’autres approches projettent d’« éditer » l’ADN (c’est-à-dire de modifier l’endroit spécifique de la mutation) sans le couper, ce qui diminue l’occurrence des modifications hors cible (autres gènes) qui sont sources d’inquiétude.6 Par exemple, l’enzyme ADAR (adenosine deaminase acting on RNA) déaminase transforme l’adénosine en inosine, ce qui permet de générer des variants non pathogènes.7
Quels que soient les moyens d’édition, l’objectif ultime de la thérapie génique est de corriger la mutation dans des cellules souches, les reprogrammer en cellules basales respiratoires afin de les réensemencer dans les voies respiratoires, pour qu’elles se différencient en cellules épithéliales.8 De nombreux obstacles restent à surmonter pour cette thérapie dite cellulaire, notamment pour réunir les conditions nécessaires à la greffe de telles cellules basales dans leur niche spécifique.
Thérapie de transcrit avec de l’ARNm chimiquement modifié
Les thérapies de transcrit visent l’ARN messager (ARNm) dans le cytoplasme, ce qui permet de s’affranchir de l’étape de translocation nucléaire. Le succès des vaccins à ARNm pendant la pandémie de Covid- 19 a marqué un tournant dans le domaine. Les stratégies actuelles emploient de l’ARNm chimiquement modifié afin, d’une part, de limiter les réactions immunitaires dues à la détection de matériel génétique exogène et, d’autre part, de stabiliser la séquence de nucléosides.9 Les essais cliniques de phase II n’ont pas montré d’efficacité clinique. De nouveaux essais ont débuté, visant à maximiser la quantité de protéines CFTR matures à la membrane plasmique.10 Surtout, des approches d’édition de l’ARNm, utilisant la technologie CRISPR associée à la nucléase Cas13, encore au stade préclinique, visent à corriger spécifiquement les bases d’ARN mutées.11
Thérapie de translecture « anti-stop »
Les mutations non-sens sont caractérisées par la présence d’un PTC – UAA, UAG ou UGA. Elles sont associées à des phénotypes plus sévères, qui se manifestent dès l’enfance.12 La présence de ces PTC conduit à la dégradation des transcrits mutés : si, malgré tout, il en persiste un peu pour initier la synthèse, la fabrication d’une protéine tronquée sera éliminée. Des molécules visant à induire des « infidélités de lecture » du code génétique permettent la synthèse d’une néoprotéine. Si l’acide aminé inséré n’affecte pas la structure de la protéine, celle-ci est alors fonctionnelle. Synthétisées en nombre suffisant, les protéines peuvent permettre la restauration de la fonction CFTR à l’échelle du tissu et de l’organisme. Les antibiotiques, comme la gentamicine, ou leurs dérivés moins toxiques, comme ELX- 02, modifient la conformation du ribosome, permettant la fixation d’ARNt, facilitant ainsi les erreurs de lecture, mais une étude de phase II n’a pas confirmé le bénéfice clinique.
De nouvelles molécules sont en développement préclinique.13 Certaines, comme des ARN de transfert (qui amènent les acides aminés sur site pour la synthèse protéique), pourraient aussi court-circuiter ce signal stop. Ces ARNt « artificiels » n’établissent que deux liaisons codon-anticodon sur trois mais peuvent néanmoins se fixer sur l’ARN, ignorer le codon stop, insérer un acide aminé dans la chaîne peptidique et permettre ainsi la poursuite de la traduction.14
Oligonucléotides antisens à l’étude
Cette stratégie est très prometteuse. Les oligonucléotides antisens (ASOs) synthétiques sont des petits fragments d’ADN conçus pour se lier spécifiquement à l’ARNm cible.15 Leur petite taille permet leur passage à travers les mucines, ce qui évite l’utilisation de véhicule d’administration, défaut majeur des thérapies à ADN ou ARN. Les ASOs bloquent la reconnaissance des mutations d’épissage. Cette stratégie s’est déjà avérée efficace dans l’amyotrophie musculaire spinale et la dystrophie musculaire de Duchenne. Dans la mucoviscidose, des résultats in vitro encourageants ont été obtenus, notamment sur des cellules respiratoires primaires portant le variant 3849 + 10kb C>T. Un essai clinique chez les patients porteurs de cette mutation 3849 + 10 kb C>T a débuté.16
Voies de recherche en défrichage
L’efficacité clinique des modulateurs de CFTR ouvre la voie aux stratégies d’édition d’ARN et d’ADN. Ces avancées ne seront certainement bientôt plus de la science-fiction. Elles illustrent les interactions fortes entre les laboratoires académiques, les industries pharmaceutiques, les associations et les autorités de santé. Cette révolution en marche permet d’envisager que la mucoviscidose ne soit plus une maladie mortelle.
2. Fajac I, Sermet I. Therapeutic approaches for patients with cystic fibrosis not eligible for current CFTR modulators. Cells 2021;10:2793.
3. Fajac I, Sermet-Gaudelus I. Emerging medicines to improve the basic defect in cystic fibrosis. Expert Opin Emerg Drugs 2022;27:229-39.
4. Griesenbach U, McLachlan G, Owaki T, et al. Validation of recombinant Sendai virus in a non-natural host model. Gene Ther 2011;18:182-8.
5. Li T, Yang Y, Qi H, et al. CRISPR/Cas9 therapeutics: Progress and prospects. Signal Transduct Target Ther 2023;8(1):36.
6. Sousa AA, Hemez C, Lei L, et al. Systematic optimization of prime editing for the efficient functional correction of CFTR F508del in human airway epithelial cells. Nat Biomed Eng 2025;9(1):7-21.
7. Gaudelli NM, Komor AC, Rees HA, et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017;551:464-71.
8. Hayes D, Kopp BT, Hill CL, et al. Cell therapy for cystic fibrosis lung disease: Regenerative basal cell amplification. Stem Cells Transl Med 2019;8:225-35.
9. Robinson E, MacDonald KD, Slaughter K, et al. Lipid nanoparticle-delivered chemically modified mRNA restores chloride secretion in cystic fibrosis. Mol Ther 2018;26:2034-46.
10. Rowe SM, Zuckerman JB, Dorgan D, et al. Inhaled mRNA therapy for treatment of cystic fibrosis: Interim results of a randomized, double-blind, placebo-controlled phase 1/2 clinical study. J Cyst Fibros 2023;22:656-64.
11. Cox DBT, Gootenberg JS, Abudayyeh OO, et al. RNA editing with CRISPR-Cas13. Science 2017;358:1019-27.
12. Orenti A, Pranke I, Faucon C, et al. Nonsense mutations accelerate lung disease and decrease survival of cystic fibrosis children. J Cyst Fibros 2023;22(6):1070-9.
13. Djumagulov M, Demeshkina N, Jenner L, et al. Accuracy mechanism of eukaryotic ribosome translocation. Nature 2021;600:543-6.
14. Albers S, Allen EC, Bharti N, et al Engineered tRNAs suppress nonsense mutations in cells and in vivo. Nature 2023;618:842-8.
15. Levin AA. Treating disease at the RNA level with oligonucleotides. New England Journal of Medicine 2019;380:57-70.
16. Oren YS, Irony-Tur Sinai M, Golec A, et al. Antisense oligonucleotide-based drug development for cystic fibrosis patients carrying the 3849+10 kb C-to-T splicing mutation. J Cyst Fibros 2021;20:865-75.