Bulles et érosions post-bulleuses
En période néonatale, de telles lésions peuvent révéler la fragilité cutanée d’une épidermolyse bulleuse héréditaire. En cause : les mutations dans des gènes codant des protéines de l’hémidesmosome (structure qui assure la cohésion entre derme et épiderme). Il faut éviter l’application de tout adhésif risquant de majorer le décollement cutané. La fragilité cutanée est constatée dès la naissance. Bulles et érosions post-bulleuses prédominent aux zones de frottement et sont souvent accompagnées d’une atteinte muqueuse. Certains cas, moins sévères, se révèlent plus tardivement par des bulles des pieds et talons lors de la marche (fig. 1). La gravité est très variable, allant de formes localisées permettant une vie quasi normale à d’autres rapidement létales. Si la maladie est sévère, l’étendue des plaies, l’atteinte muqueuse, les cicatrisations à répétition sont sources de complications systémiques : dénutrition, douleur, amylose, carcinomes épidermoïdes cutanés…
Ichtyoses et troubles de la kératinisation
Les ichtyoses héréditaires sont dues à des mutations dans des gènes impliqués dans la formation de la barrière cutanée (plus de 50). Elles sont caractérisées par une hyperkératose et une accumulation excessive de squames sur la surface de la peau, et parfois d’une érythrodermie. Des manifestations extracutanées sont possibles, surtout neurologiques et ophtalmologiques. Il faut être attentif au développement psychomoteur de l’enfant et au risque de carence en vitamine D.
L’érythrodermie congénitale ichtyosiforme bulleuse est extrêmement rare (aspect « d’enfant ébouillanté » puis érythrodermie couverte de larges décollements cutanés).
L’ichtyose vulgaire apparaît rapidement et persiste toute la vie. L’atteinte prédomine aux faces d’extension des membres et respecte les grands plis ; les squames sont fines et le prurit modéré. Elle s’améliore l’été et s’aggrave l’hiver.
L’ichtyose récessive liée à l’X se manifeste rapidement après la naissance, avec des lésions symétriques sur les membres et les faces latérales du visage et du tronc. Les grands plis sont moins respectés que dans l’ichtyose vulgaire et les squames sont de plus grande taille. Les formes extensives donnent un aspect « sale » au tégument. Les femmes, souvent transmettrices, ont pour seule lésion une sécheresse cutanée de la face antérieure des jambes.
Le traitement des ichtyoses, symptomatique, repose sur les soins associant bains/douches et l’application d’émollients +/- enrichis en substances kératolytiques. La HAS a élaboré récemment des recos à lire ici.
Hyperpigmentations
Les taches café au lait (TCL) sont des macules uniformément pigmentées de couleur brun clair/noir, rondes ou ovales. Elles peuvent être congénitales ou apparaître dans les premières années de vie. Leur taille, variable, augmente avec la croissance. Si une TCL solitaire est une « tache de naissance » banale et physiologique du nouveau-né, de nombreuses affections génétiques comportent des TCL multiples.
En pratique, seules quelques-unes doivent être recherchées de prime abord par le praticien.
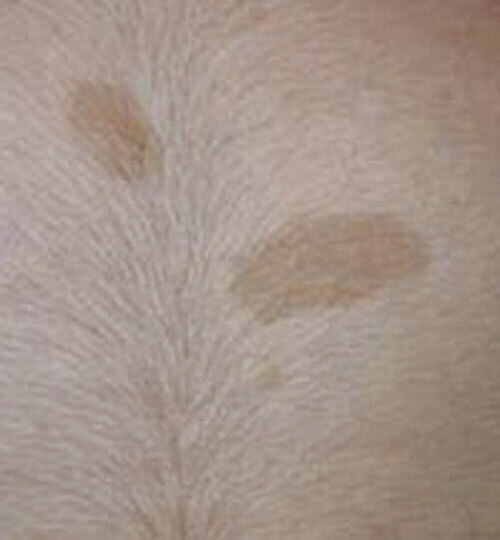
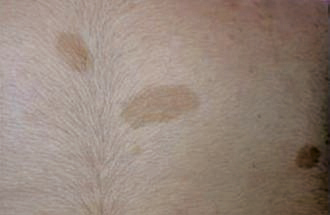
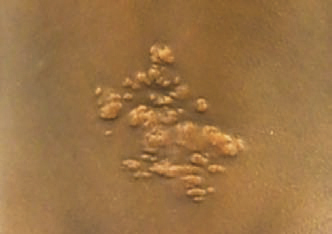
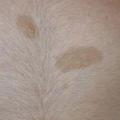
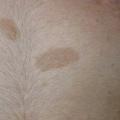
La neurofibromatose de type 1 (NF1), la plus fréquente, est le principal défi diagnostique, surtout en l’absence d’autre anomalie clinique ou d’antécédent familial (survenue de novo chez 50 % des patients). Son expressivité inter- et intrafamiliale est variable. Ces dernières années ont été marquées par des avancées significatives dans le diagnostic, le suivi et la prise en charge. Les critères diagnostiques de NF1 ont été révisés récemment (tableau). Critère le plus évident : plus de 6 taches café au lait (TCL) > 5 mm avant la puberté ou > 1,5 cm après (fig. 2). Les lentigines (TCL millimétriques ; fig. 3), fréquentes, prédominent dans les régions axillaire et inguinale.
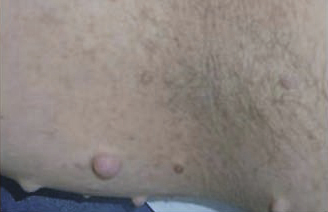
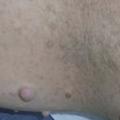
La NF1 prédispose au développement de tumeurs, bénignes ou malignes, justifiant certains dépistages et une surveillance. Les tumeurs malignes des gaines nerveuses sont les plus redoutées. Les neurofibromes, tumeurs cutanées papulo-nodulaires, surviennent surtout au moment de la puberté (fig. 4). Dans les neurofibromes plexiformes inopérables de l’enfant, un nouveau médicament est disponible en France (sélumétinib).
Chez ces enfants, on surveille la croissance staturo-pondérale, la PA, les apprentissages, l’acuité et le champ visuel (gliome des voies optiques), la statique (scoliose par dysplasies vertébrales), l’audition et des signes orientant vers une puberté précoce.
Le syndrome de Legius, principal diagnostic différentiel de la NF1, associe des TCL et des lentigines axillaires, mais sans risque de neurofibrome ni de gliome optique.
Les autres affections génétiques associées à des TCL multiples sont généralement révélées par des manifestations extracutanées, en particulier neurologiques. Devant une hyperpigmentation linéaire et segmentaire (d’allure banale), il ne faut pas méconnaître les 2 affections suivantes :
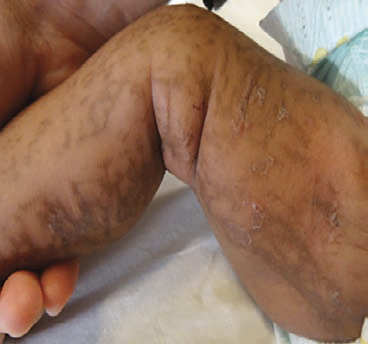
- Dans l’incontinentia pigmenti, atteignant surtout les nouveau-nés de sexe féminin (transmission liée à l’X), les lésions évoluent souvent en 4 stades : vésiculo-bulleux, verruqueux hyperkératosique, hyperpigmenté (fig. 5) puis atrophique et hypopigmenté. Un avis dermatologique, neurologique et ophtalmologique urgent s’impose (risque de décollement de rétine et/ou de crise convulsive).
- Le syndrome de McCune-Albright associe TCL de grande taille, à bords irréguliers (fig. 6), dysplasie fibreuse polyostosique (douleurs + hypertrophie volontiers asymétrique de la voûte crânienne et du massif facial) et manifestations endocriniennes (puberté précoce et aussi hyperthyroïdie, acromégalie, syndrome de Cushing…).
Hypopigmentations
Les albinismes oculo-cutanés sont caractérisés par une hypopigmentation de degré variable de la peau, des phanères et des yeux. La prise en charge ophtalmologique est essentielle (photophobie, nystagmus). Des troubles de la coagulation (hématomes, épistaxis) et des infections à répétition doivent être recherchés. Les principales complications dermatologiques sont les kératoses actiniques , les carcinomes épidermoïdes, basocellulaires et les mélanomes (beaucoup plus rares). Un suivi dermatologique est donc recommandé au moins 2 fois par an. La photoprotection cutanée et oculaire est essentielle et s’accompagne d’une substitution en vitamine D à vie. Le développement psychomoteur de l’enfant peut être soutenu par un psychomotricien. Un suivi psychologique peut être proposé. Les récentes recos de la HAS sont abordées ici.
La sclérose tubéreuse de Bourneville se manifeste par la survenue de tumeurs bénignes (hamartomes) dans divers tissus. La peau, le cerveau, les reins, les yeux et le cœur sont les organes à surveiller. Les macules hypopigmentées apparaissent dès les premiers mois de vie (au moins 3 et de taille > 0,5 cm pour évoquer le diagnostic). Leur aspect en « feuille de sorbier » est caractéristique, allongé, ovalaire, polygonal, asymétrique, touchant le tronc et les fesses. La plaque fibreuse du front et celle en peau de chagrin lombaire (fig. 7) sont plus tardives. Les angiofibromes faciaux, très souvent confondus avec de l’acné juvénile, sont des papulo-nodules érythémateux télangiectasiques des régions périnasale et jugale ; leur précocité rectifie le diagnostic.
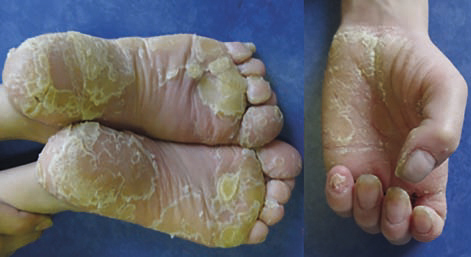
Kératodermies palmoplantaires (KPP)
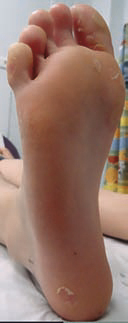
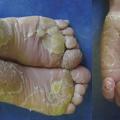
L’épaississement de la peau des paumes et des plantes (fig. 8) retentit sur la marche (souvent douloureuse) et la motricité fine (écriture). Dans certaines situations, elle peut révéler, associée à une alopécie, une cardiomyopathie sévère.
Pathologies du tissu conjonctif
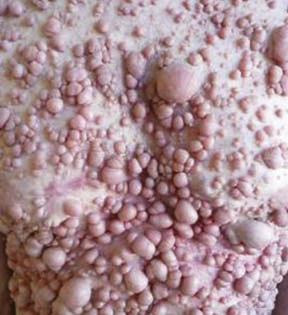
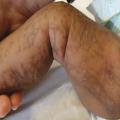
Syndrome d’Ehlers-Danlos (dysplasie des fibres collagènes) : dans la forme hypermobile (la plus fréquente), la peau est fine, hyperlaxe (fig. 9), avec des hématomes, des cicatrices atrophiques. S’y associe une hyperlaxité des grandes et petites articulations (fig. 10). Chez certains patients, la discrétion de l’atteinte cutanée retarde le diagnostic. Signes évocateurs : fatigue, douleurs diffuses, fragilité cutanée... Le blocage des articulations retentit sur l’écriture et les gestes fins. Contrairement à la forme vasculaire, il n’y a pas d’atteinte des gros vaisseaux pouvant mettre en jeu le pronostic vital.
Dans la maladie de Marfan (dysplasie des fibres élastiques), l’atteinte est musculosquelettique (grandes taille et envergure, déformation du thorax, hyperlaxité ligamentaire), oculaire (ectopie du cristallin) ou cardiovasculaire (dilatation aortique). L’examen dermatologique révèle des vergetures, des hernies (inguinales), une arachnodactylie (doigts longs et fins).
Pathologies dysimmunitaires
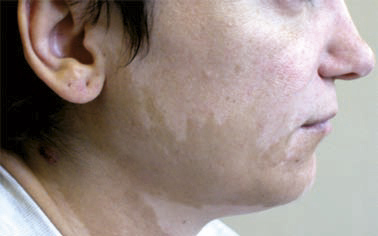
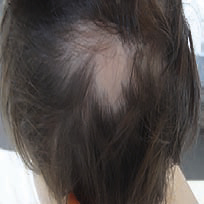
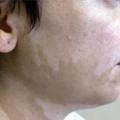
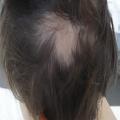
Une pelade (fig. 11) ou un vitiligo accompagnent parfois une polyendocrinopathie auto-immune. Des lésions eczématiformes ou psoriasiformes atypiques sont souvent associées.
Points clés
➜ Le diagnostic est clinique : on examine le tégument et les muqueuses, mais aussi les annexes.
➜ Le caractère affichant des lésions retentit sur les relations sociales, affectant la qualité de vie des patients et de leurs familles ; certaines peuvent mettre en jeu le pronostic vital.
➜ Une à trois taches café au lait (TCL) isolées chez l’enfant d’âge scolaire ne sont pas pathologiques. En cas de TCL multiples, le risque de NF1 est estimé à 30 % et augmente à 75 % si le nombre est ≥ 6.
Fertitta L, Wolkenstein P. Neurofibromatose de type 1 : des avancées récentes. Rev Prat 2025;75(3);321-4.