Le terme « sarcome » regroupe une large famille de tumeurs développées aux dépens des cellules du tissu conjonctif, comportant deux sous-familles, les sarcomes des tissus mous (STM, environ 80 % des cas) et les sarcomes osseux (SO, environ 20 % des cas). Il s’agit d’un groupe hétérogène de tumeurs, avec actuellement plus de 150 entités distinctes identifiées (sous-types histologiques).1 Cette hétérogénéité se traduit en matière de localisation et de pronostic évolutif :
- même si les membres représentent environ 60 % des localisations des tumeurs primitives, les sarcomes peuvent se développer dans n’importe quel site anatomique. En effet, on retrouve des sarcomes primitifs cutanés, superficiels (c’est-à-dire sous-cutanés), profonds, viscéraux, exceptionnellement encéphaliques ;
- parmi les entités des tumeurs conjonctives, on retrouve des tumeurs strictement bénignes (comme le lipome ou le léiomyome utérin), des tumeurs d’agressivité locale (parfois dites « à malignité intermédiaire ») comme la tumeur desmoïde ou le dermatofibrosarcome de Darier et Ferrand et des tumeurs malignes à risque métastatique, « vrais » sarcomes, comme le sarcome pléomorphe ou le léiomyosarcome.
Il s’agit de pathologies rares, nécessitant le plus souvent un traitement multimodal complexe et une prise en charge en milieu spécialisé dès le diagnostic afin de ne pas impacter négativement le pronostic des patients.
Cependant, les médecins de ville doivent être sensibilisés à ces pathologies et connaître la conduite à tenir en cas de suspicion de tumeur conjonctive afin de donner les meilleures chances aux patients.
Des pathologies rares
L’incidence des sarcomes est faible, quoique sans doute sous-évaluée. Ils peuvent survenir à tout âge et, bien que la plupart soient sporadiques, de nombreux facteurs de risque sont bien identifiés.
Incidence sous-évaluée
Les sarcomes sont des pathologies rares. Tous histotypes confondus, leur incidence « historique » était estimée à environ 4 à 6 cas pour 100 000 habitants.2
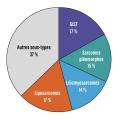
En France, dans le cadre de la structuration de la prise en charge des patients atteints de sarcomes, tous les diagnostics anatomopathologiques doivent être relus par des pathologistes experts dans le cadre du Réseau de référence en pathologie des sarcomes (RRePS). Cet enregistrement national prospectif des nouveaux cas de sarcome depuis plusieurs décennies a récemment permis de montrer que cette incidence historique est probablement sous-évaluée, l’incidence des tumeurs conjonctives malignes étant de 70,7 pour 100 000 selon les données récentes.3 Ce chiffre regroupe plusieurs dizaines d’entités différentes, de fréquence variable (quelques cas par an en France pour les formes de sarcomes les plus rares), même si la somme des incidences des quatre entités les plus fréquentes (tumeurs stromales gastro-intestinales [GIST], sarcomes pléomorphes, léiomyosarcomes et liposarcomes) représente près de deux tiers des cas (fig. 1).
Âge de survenue très variable
L’âge médian de survenue d’un sarcome se situe autour de 60 ans. Néanmoins, il faut retenir que les tumeurs conjonctives, qu’elles soient STM ou SO, à malignité intermédiaire ou véritable sarcome, peuvent se développer à tout âge. Ainsi, le réflexe de penser à un sarcome devant une masse des tissus mous ou osseuse doit être le même quel que soit l’âge du patient.
Le sex-ratio des patients atteints est équilibré.
En revanche, le spectre des sous-types rencontrés diffère en fonction des classes d’âge :
- les rhabdomyosarcomes, embryonnaires ou alvéolaires, représentent la moitié des cas de sarcomes diagnostiqués chez les enfants de moins de 15 ans.4 Une quinzaine d’autres entités rares et moins « spécifiques » de la population pédiatrique constitue la seconde moitié ;
- la population dite « adolescente/jeunes adultes » (AJA, de 15 à 25 ans) se situe entre l’épidémiologie des sarcomes pédiatriques et celle des adultes. Il s’agit en outre de l’âge de survenue moyen des deux formes de sarcomes osseux primitifs des jeunes (l’ostéosarcome et le sarcome d’Ewing) ;
- l’épidémiologie des patients adultes regroupe 150 sous-types histologiques différents, au sein desquels un petit nombre d’entités prédominent, principalement des sarcomes à génomique complexe (les léiomyosarcomes, les liposarcomes, bien différenciés ou dédifférenciés, et les sarcomes pléomorphes, indifférenciés).
Facteurs de risque spécifiques
La majorité des sarcomes, tant STM que SO, surviennent de manière sporadique, c’est-à-dire sans facteur de risque connu. Néanmoins, il existe certains facteurs de risque classiques à connaître.
Radiothérapie
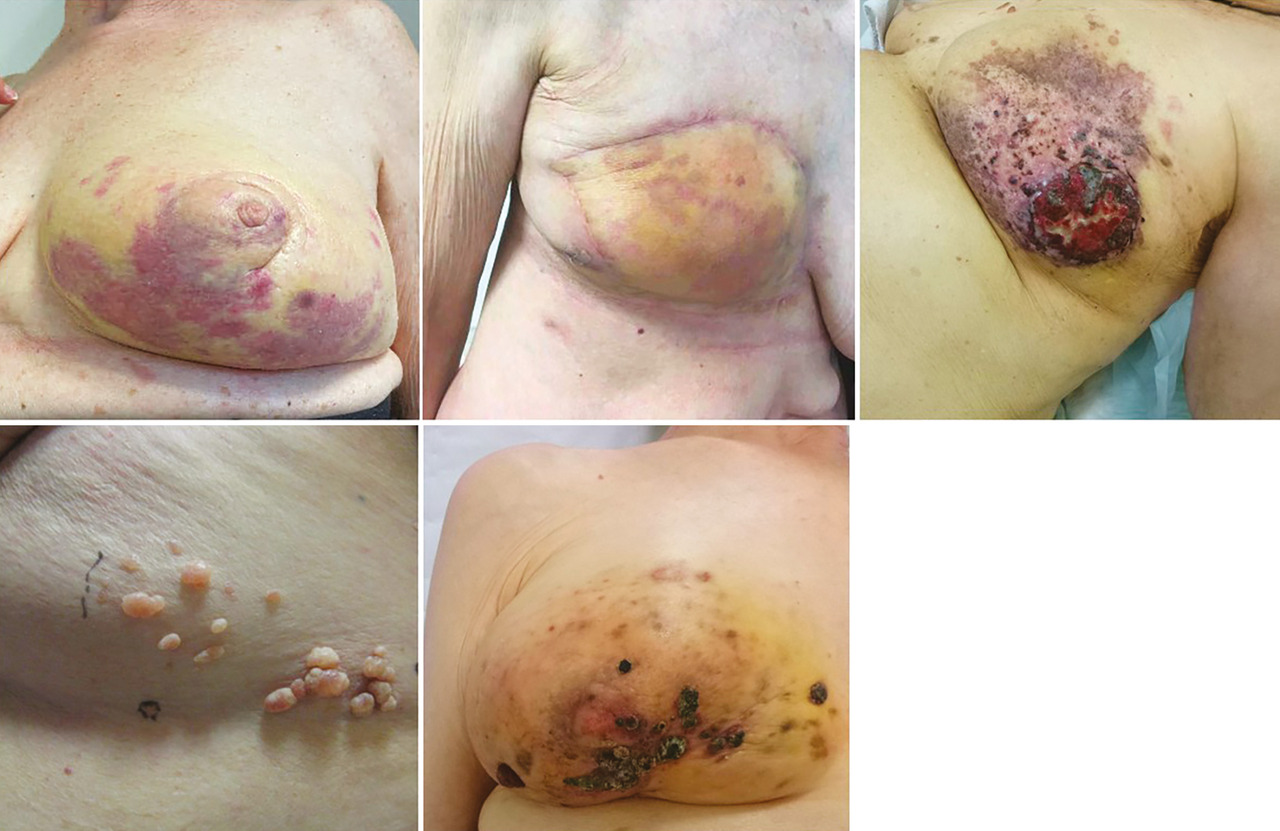
La radiothérapie est un traitement-clé du contrôle local de plusieurs types de cancer (dont les sarcomes !), soit en complément d’une chirurgie, soit en tant que traitement exclusif. Le développement d’un sarcome dans un champ précédemment irradié est une complication, rare (entre 0,02 et 0,8 %) mais grave, de toute irradiation.5 Il peut s’agir de STM ou de SO, les sous-types les plus fréquemment observés dans ces cas étant l’angiosarcome (cutané principalement), les sarcomes pléomorphes ou les ostéosarcomes. Une grande partie des sarcomes développés en territoire irradié sont des angiosarcomes mammaires. Le délai de survenue médian d’un sarcome en territoire irradié est de dix ans, mais peut être plus précoce, dès deux ans après l’irradiation.
Toute tumeur des tissus mous ou osseuse, ou toute modification cutanée dans un champ précédemment irradié est potentiellement suspecte et doit faire envisager le diagnostic de sarcome en territoire irradié (fig. 2).
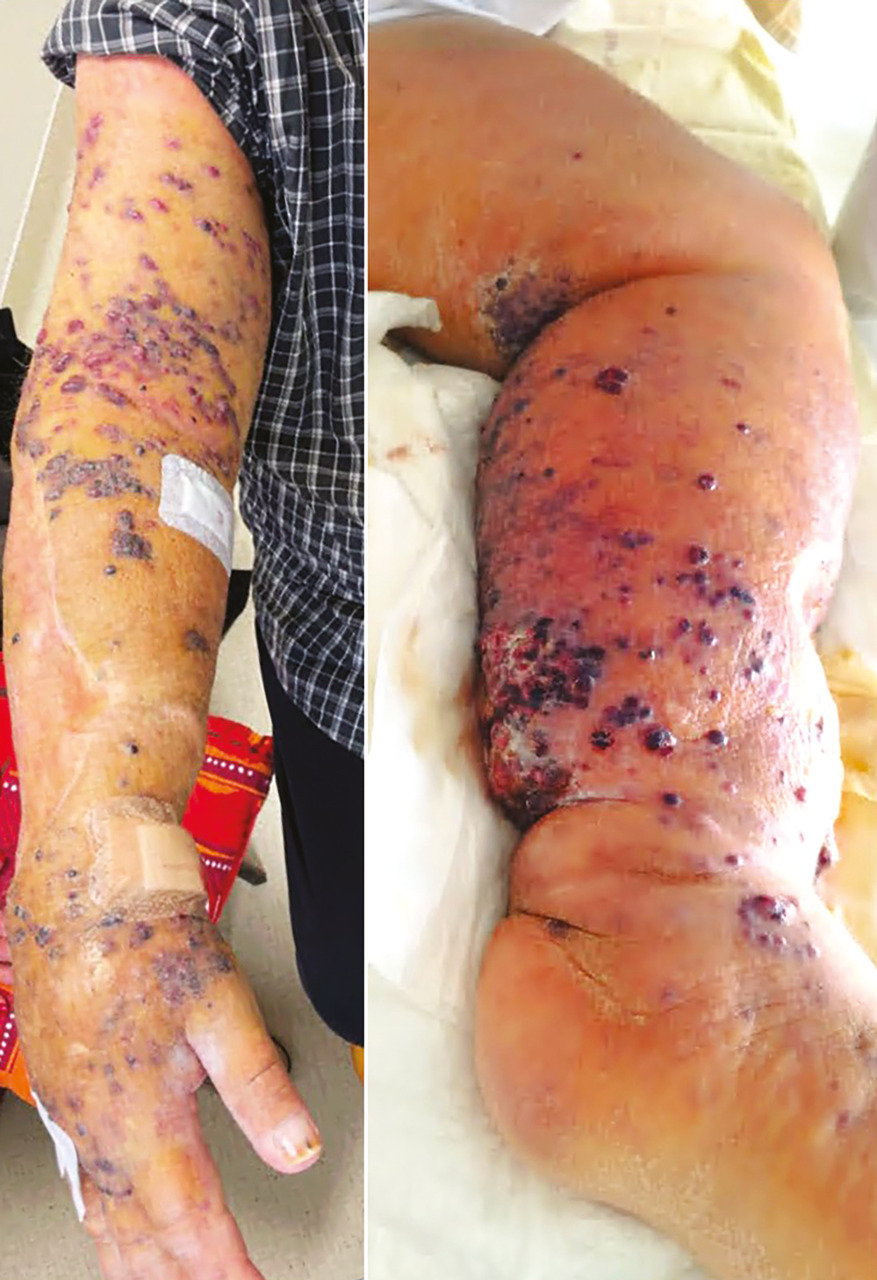
Lymphœdème chronique
Les patients porteurs d’un lymphœdème chronique, quelle qu’en soit la cause, présentent un faible risque de développer un angiosarcome cutané dans le territoire concerné (syndrome de Stewart-Treves).6 Comme pour les angiosarcomes en territoire irradié, toute modification cutanée chez un patient porteur de lymphœdème est potentiellement suspecte (fig. 3).
Prédispositions génétiques
Certaines affections génétiques peuvent prédisposer au développement de STM ou SO, parfois dès l’enfance. Certains tableaux cliniques incitent à rechercher ces prédispositions, qui impactent potentiellement les apparentés du patient du fait d’une transmission génétique possible.
Neurofibromatose de type 1
Due à une mutation du gène RB1, la neurofibromatose de type 1 est la plus fréquente des maladies autosomiques à transmission dominante, avec une incidence allant de 1 pour 2 500 à 1 pour 5 000 naissances,7 même si environ la moitié des formes de NF1 sont sporadiques (c’est-à-dire de novo et non héritées d’un parent).
Elle associe classiquement (mais de manière variable selon les individus) des taches cutanées café au lait et des neurofibromes cutanés. Certains patients présentent également des tumeurs profondes, parfois multiples, appelées neurofibromes plexiformes, ayant un risque de transformation en tumeur maligne des gaines nerveuses périphériques (MPNST, sous-type de STM) d’environ 10 % au cours de la vie .8 Toute modification de taille ou symptomatique d’un neurofibrome connu chez un patient porteur de NF1 est un signe d’alerte (lire les articles « Neurofibromatose de type 1 : des avancées récentes » dans La Revue du Praticien de mars 2025 et « Vivre avec une neurofibromatose de type 1 » dans La Revue du Praticien de mars 2024).
Syndrome de Li-Fraumeni
Causé par une mutation du gène TP53, le syndrome de Li-Fraumeni (LFS) correspond à une autre prédisposition à transmission autosomique dominante. Il est propice au développement d’un certain nombre de cancers (mammaires, cérébraux, sarcomes, surrénaliens), survenant chez des patients souvent jeunes, parfois enfants. La présence des critères de Chompret, correspondant à la survenue de cancers du spectre LFS chez des patients jeunes ou leurs apparentés, doit faire rechercher cette prédisposition. Les critères de Chompret se rapportent à la présence d’un item parmi les trois suivants :
- patient atteint d’un cancer du spectre Li-Fraumeni (sarcome des tissus mous, ostéosarcome, cancer du sein, corticosurrénalome, leucémie, carcinome bronchiolo-alvéolaire pulmonaire) avant 46 ans ET au moins un antécédent familial au premier ou second degré d’un cancer du spectre Li-Fraumeni avant 56 ans ;
- patient avec antécédent personnel de deux cancers appartenant au spectre Li-Fraumeni, dont l’un survenu avant 46 ans ;
- patient atteint d’un corticosurrénalome ou d’un carcinome des plexus choroïdes, indépendamment de l’histoire familiale.9,10
Autres affections génétiques
Outre ces deux syndromes principaux, d’autres anomalies génétiques peuvent prédisposer, de manière plus rare, au développement des sarcomes : la mutation du gène RB1, prédisposant au développement de STM et SO ;11 la mutation APC (responsable de la polypose adénomateuse familiale), parfois associée au développement de tumeurs desmoïdes abdominales (syndrome de Gardner) ;12 ou même le syndrome de Lynch (regroupant plusieurs anomalies génétiques typiquement associées aux cancers digestifs), lié à certaines formes de sarcomes pléomorphes.13
Exposition à des toxiques
L’exposition au chlorure de vinyle (utilisé notamment dans l’industrie plastique) prédispose au développement d’angiosarcome hépatique.14 Il s’agit d’une cause classiquement décrite, mais rarissime en pratique de nos jours.
Centralisation de la prise en charge : nécessité de l’adressage des patients au centre NetSarc+
En France, la prise en charge des patients atteints de sarcome est structurée au sein du réseau multidisciplinaire NetSarc+. Ce réseau d’expertise est né en 2019 de la fusion de trois réseaux complémentaires préexistants :
- le réseau de relecture anatomopathologique RRePS, permettant le diagnostic précis des tumeurs conjonctives. Il s’agit d’une étape majeure de la prise en charge des patients, le taux de discordance entre la relecture par un pathologiste spécialisé et un pathologiste non expert étant de plus de 40 % ;15
- le réseau clinique NetSarc, constitué d’équipes médicales régionales de cliniciens référents pour la prise en charge des patients ;
- le réseau RésOS, regroupant à la fois les pathologistes et les cliniciens spécialisés dans la prise en charge des sarcomes osseux.
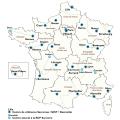
Ce réseau s’articule autour d’un centre de référence multisite (centre Léon-Bérard à Lyon, institut Bergonié à Bordeaux, Gustave-Roussy à Villejuif) et de 22 centres de compétence régionaux dont la liste et les coordonnées sont accessibles sur le site expertise sarcome (https ://expertisesarcome.org/centres-experts-par-region/) [fig. 4].
Les patients atteints de sarcome doivent être pris en charge dès la suspicion diagnostique dans les centres NetSarc+. Outre les pièges diagnostiques, détaillés dans l’article suivant, qui peuvent impacter négativement le début de la prise en charge, il est scientifiquement prouvé que le pronostic des patients est meilleur lorsqu’ils sont traités dans les centres spécialisés.
Dans plusieurs publications au cours des vingt dernières années, le Groupe sarcome français–Groupe d’étude des tumeurs osseuses a en effet montré que la survie des patients atteints de sarcome est meilleure lorsqu’ils sont traités dans les centres spécialisés.16 - 18 Cela s’explique par la meilleure adhésion aux recommandations de prise en charge diagnostique et thérapeutique, et surtout par la qualité de la chirurgie carcinologique, facteur pronostique majeur des sarcomes localisés, qui doit être réalisée par des chirurgiens experts.
Attention aux idées reçues !
Il est nécessaire de rétablir la vérité concernant certaines idées reçues sur les sarcomes (encadré). Un message simple est à retenir pour la pratique quotidienne : « Toute masse des tissus mous ou osseuse, quel que soit l’âge du patient, est un sarcome jusqu’à preuve du contraire et nécessite une discussion dans une réunion de concertation pluridisciplinaire NetSarc+ avant tout geste. »
Quelques idées reçues SUR les sarcomes
Les sarcomes sont une pathologie de l’enfant.
→ FAUX, l’âge médian au diagnostic est de 60 ans.
Les sarcomes sont essentiellement osseux.
→ FAUX, 80 % des sarcomes se développent en dehors du tissu osseux.
Les sarcomes surviennent essentiellement dans un contexte génétique connu.
→ FAUX, les formes sporadiques représentent plus de 95 % des cas.
Les sarcomes ont un effroyable pronostic.
→ FAUX, la survie à cinq ans est estimée à 60 % (largement influencée par la prise en charge en centre spécialisé dès la suspicion diagnostique).
2. Ng VY, Scharschmidt TJ, Mayerson JL, et al. Incidence and survival in sarcoma in the United States: A focus on musculoskeletal lesions. Anticancer Res 2013; 33(6):2597‑604.
3. de Pinieux G, Karanian M, Le Loarer F, et al. Nationwide incidence of sarcomas and connective tissue tumors of intermediate malignancy over four years using an expert pathology review network. PLoS One 2021;16(2):e0246958.
4. Loeb DM, Thornton K, Shokek O. Pediatric soft tissue sarcomas. Surg Clin North Am 2008;88(3):615‑27.
5. Snow A, Ring A, Struycken L, et al. Incidence of radiation induced sarcoma attributable to radiotherapy in adults: A retrospective cohort study in the SEER cancer registries across 17 primary tumor sites. Cancer Epidemiol 2021;70:101857.
6. Stewart FW, Treves N. Lymphangiosarcoma in postmastectomy lymphedema: A report of six cases in elephantiasis chirurgica. Cancer 1948;1:64‑81.
7. Huson SM, Compston DA, Clark P, et al. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. I. Prevalence, fitness, mutation rate, and effect of parental transmission on severity. J Med Genet 1989;26(11):704‑11.
8. McGaughran JM, Harris DI, Donnai D, et al. A clinical study of type 1 neurofibromatosis in north west England. J Med Genet 1999;36(3):197‑203.
9. Bougeard G, Renaux-Petel M, Flaman JM, et al. Revisiting Li-Fraumeni syndrome from TP53 mutation carriers. J Clin Oncol 2015;33(21):2345-52.
10. Tinat J, Bougeard G, Baert-Desurmont S, et al. 2009 version of the Chompret criteria for Li Fraumeni syndrome. J Clin Oncol 2009;27(26):e108-9.
11. Kleinerman RA, Schonfeld SJ, Tucker MA. Sarcomas in hereditary retinoblastoma. Clin Sarcoma Res 2012;2(1):15.
12. Galiatsatos P, Foulkes WD. Familial adenomatous polyposis. Am J Gastroenterol 2006;101(2):385‑98.
13. Poumeaud F, Valentin T, Fares N, et al. Sarcomas developed in patients with Lynch Syndrome are enriched in pleomorphic soft-tissue sarcomas and are sensitive to immunotherapy. Eur J Cancer 2025;216:115196.
14. Mundt KA, Dell LD, Crawford L, et al. Quantitative estimated exposure to vinyl chloride and risk of angiosarcoma of the liver and hepatocellular cancer in the US industry-wide vinyl chloride cohort: Mortality update through 2013. Occup Environ Med 2017;74(10):709‑16.
15. Ray-Coquard I, Montesco MC, Coindre JM, et al. Sarcoma: Concordance between initial diagnosis and centralized expert review in a population-based study within three European regions. Ann Oncol 2012;23(9):2442‑9.
16. Ray-Coquard I, Thiesse P, Ranchere-Vince D, et al. Conformity to clinical practice guidelines, multidisciplinary management and outcome of treatment for soft tissue sarcomas. Ann Oncol 2004;15(2):307‑15.
17. Derbel O, Heudel PE, Cropet C, et al. Survival impact of centralization and clinical guidelines for soft tissue sarcoma (A prospective and exhaustive population-based cohort). PloS One 2017;12(2):e0158406.
18. Blay JY, Honore C, Stoeckle E, et al. Surgery in reference centers improves survival of sarcoma patients: A nationwide study. Ann Oncol 2019;30(7):1143-53.
 Encadrés
Encadrés