La fœtoscopie couplée à l’usage du laser ainsi que le développement de drains pleuro-amniotiques ont changé le pronostic d’un certain nombre de pathologies fœtales thoraciques. Les procédures employées sont dites mini-invasives sur le versant maternel mais sont à risque pour l’enfant à naître. Elles ne doivent être considérées que si la pathologie est grave et menace la survie fœtale ; la localisation thoracique des pathologies expose en effet à un risque majeur de compression médiastinale et donc d’insuffisance cardiaque congestive. Le traitement de la hernie de coupole diaphragmatique congénitale est détaillé page 646.²
Épanchement pleural compressif
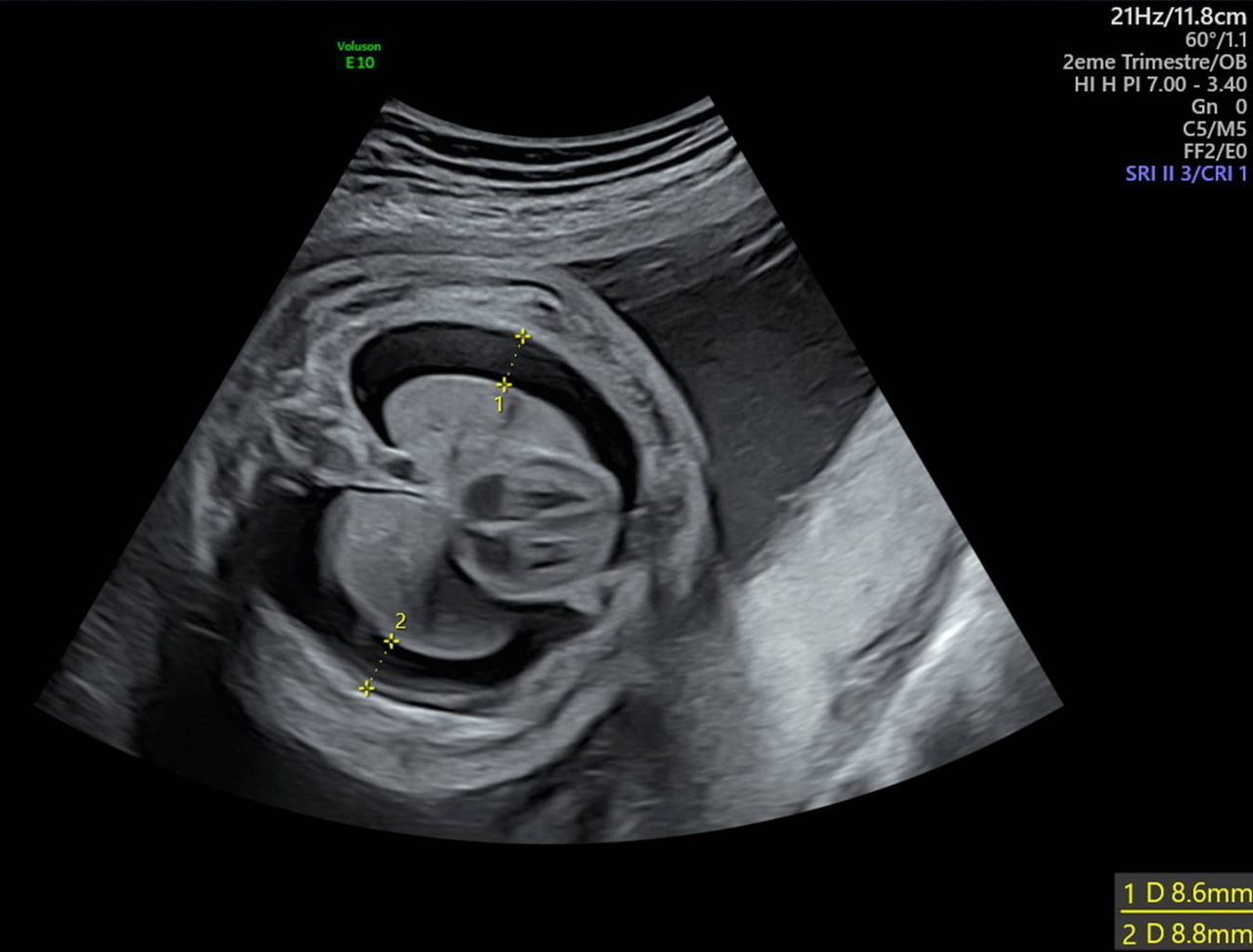
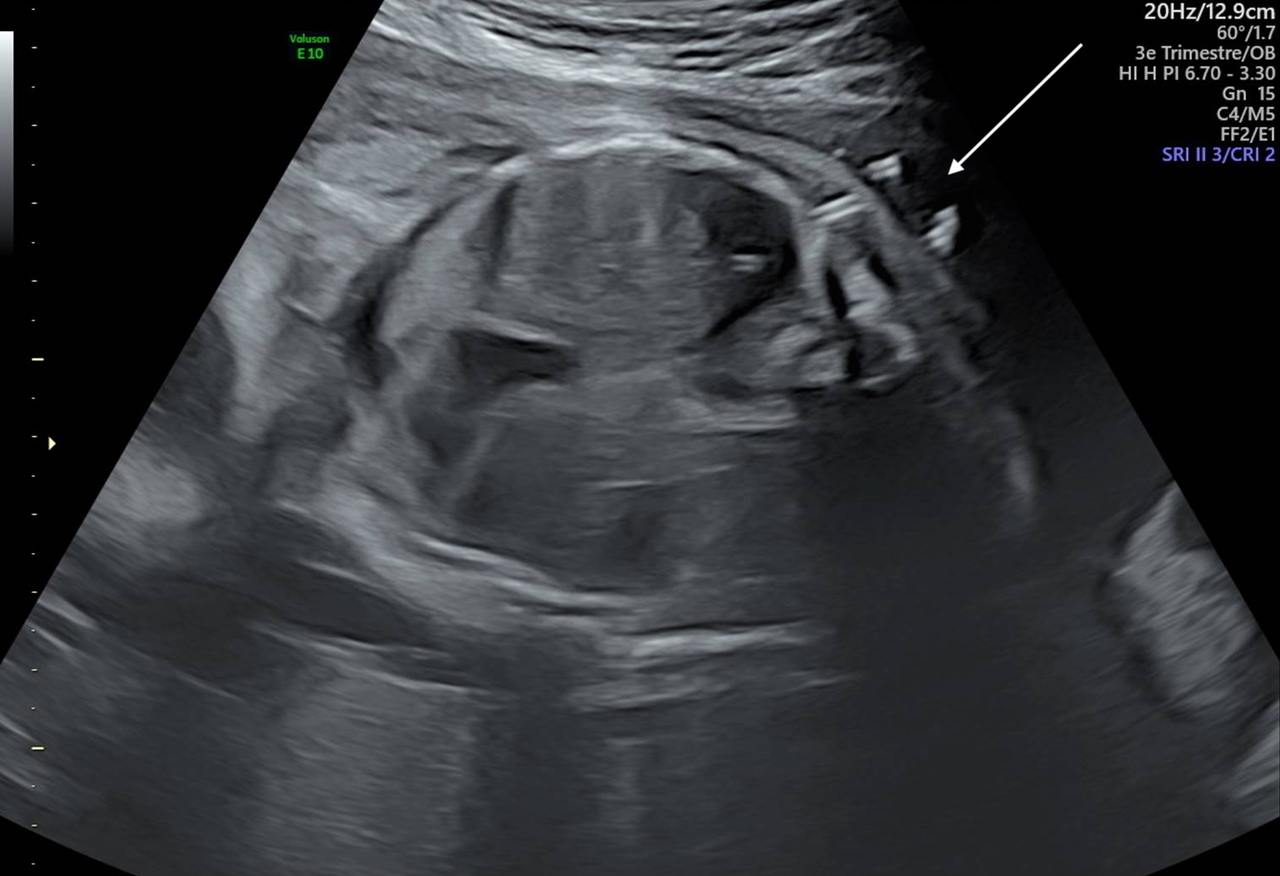
L’épanchement pleural, ou hydrothorax, est une pathologie rare qui correspond à une accumulation de liquide dans l’espace pleural chez le fœtus. Son incidence est d’environ 1/10 000 à 1/15 000 grossesses.1 L’épanchement peut être uni- ou bilatéral et apparaît, en échographie de dépistage, comme une zone anéchogène entre les poumons et la paroi thoracique (fig. 1). L’hydrothorax doit être différencié de l’épanchement péricardique dont les causes et la prise en charge ne sont pas les mêmes. Le chylothorax primitif est la première cause d’épanchement pleural chez le fœtus (65 %). Il correspond à une accumulation de lymphe dans l’espace pleural2 et peut être dû à une fuite du canal thoracique, à une surproduction de lymphe, à une dysfonction du drainage lymphatique ou encore à une anomalie de développement des canaux lymphatiques. Il s’agit d’un diagnostic d’exclusion, qui est évoqué lorsque le reste du bilan est négatif (génétique, chromosomique, infectieux). En cas d’hydrothorax compressif, les conséquences peuvent être sévères et mettre en jeu la survie du fœtus. La diminution de l’espace thoracique entraîne une perturbation du développement pulmonaire responsable d’une hypoplasie pulmonaire. L’effet de masse de l’épanchement peut également provoquer des troubles du rythme cardiaque et plus fréquemment une insuffisance cardiaque, par anomalie du retour veineux pulmonaire, pouvant aller jusqu’à l’anasarque fœtale. La présence d’une anasarque, d’une inversion des coupoles diaphragmatiques ou simplement d’un hydramnios débutant doit faire poser le diagnostic d’hydrothorax compressif et l’indication d’un drainage en urgence. Celui-ci permet de diminuer la mortalité périnatale en réduisant la compression médiastinale responsable de l’hydramnios, voire d’une anasarque fœtale.3 La pose d’un drain de dérivation pleuro-amniotique est un geste techniquement difficile qui doit être réalisé par un opérateur entraîné au sein d’un centre pluridisciplinaire de diagnostic prénatal (CPDPN). Le risque de rupture de la poche des eaux lié au geste est en moyenne autour de 20 %.3 Le risque de déplacement du drain et la possibilité d’avoir à effectuer plusieurs poses doivent aussi être expliqués. La pose se fait sous contrôle échographique continu et sous anesthésie locale ou locorégionale en fonction de la préférence de la patiente et du caractère uni- ou bilatéral de l’épanchement. Une sédation fœtale est réalisée avant la pose du drain, par injection, dans la cuisse ou dans le cordon ombilical en fonction de son accessibilité, de sufentanil et d’atracurium. La pression du liquide pleural constatée au moment du drainage permet également d’orienter le diagnostic. Une pression importante est davantage en faveur d’un hydrothorax primitif ; à l’inverse, si le liquide n’est pas sous pression, un diagnostic associé comme une maladie métabolique est plus probable. Si un hydramnios existe, il est drainé dans le même temps que la pose de drain, en gardant à l’esprit que la fonte d’une anasarque s’accompagne d’une augmentation de la quantité de liquide amniotique. Si le ou les drains sont bien posés (fig. 2), à J1 de la pose, les poumons doivent être revenus à la paroi.
À la naissance, l’enfant doit bénéficier d’une évaluation pédiatrique spécialisée, idéalement en unité de réanimation néonatale, en raison du risque de détresse respiratoire, de récidive de l’épanchement pleural ou de persistance d’un chylothorax réfractaire. Le suivi initial repose sur l’examen clinique, la surveillance respiratoire, la radiographie thoracique et l’échographie pleurale.
En cas d’épanchement persistant ou récidivant, un drainage pleural peut être nécessaire. La prise en charge associe alors des mesures nutritionnelles spécifiques (régime pauvre en graisses et riche en triglycérides à chaîne moyenne), parfois prolongées et une surveillance de la croissance, de l’état nutritionnel et du retentissement respiratoire. L’administration de somatostatine, voire un recours à la chirurgie (ligature du canal thoracique), peuvent parfois être discutés. À distance, un suivi pédiatrique pneumologique est souhaitable afin de s’assurer de l’absence de récidive et du bon développement respiratoire.
Malformation adénomatoïde kystique pulmonaire compressive
Les malformations adénomatoïdes kystiques pulmonaires (MAKP) sont les malformations pulmonaires les plus fréquentes. Elles sont le plus souvent unilatérales et unilobaires, avec une prédilection pour les lobes inférieurs. Elles sont isolées dans la grande majorité des cas. Les MAKP ont une incidence de 1 sur 7 200 naissances vivantes.4 Les dernières hypothèses suggèrent une asynchronie dans la différenciation épithélio-mésenchymateuse pendant la phase canaliculaire du développement pulmonaire, ce qui conduit à des altérations de la prolifération cellulaire et de l’apoptose. La classification simplifiée d’Adzick,5 fondée sur les données échographiques, permet de classifier les lésions en trois groupes : kystique (kystes de plus de 5 mm de diamètre), solide (kystes de moins de 5 mm de diamètre) ou mixte. Le taux de survie associé à ces malformations est de 90 %.
Une lésion peut disparaître à l’échographie du fait de modifications de l’échogénicité de la lésion qui reste toujours présente. Dans tous les cas, une surveillance, à la recherche de signes de mauvaise tolérance, est à mettre en place :
soit la lésion est découverte de façon systématique et la fréquence des échographies de surveillance est adaptée à la taille de la lésion ; les lésions kystiques compressives peuvent être surveillées parfois de façon hebdomadaire ;
soit la lésion est d’emblée compressive et un drainage est indiqué.
Il n’y a jamais d’indication à simplement ponctionner un ou plusieurs kystes, le liquide ponctionné se reconstituant dans les vingt-quatre heures. Le seul drainage efficace est la mise en place d’un drain de dérivation kysto-amniotique. Ce geste permet également d’effondrer la paroi de plusieurs kystes et de les drainer dans le même temps. Le bon positionnement du cathéter est contrôlé de façon hebdomadaire, car il peut se boucher ou migrer. La pose précoce (avant 21 semaines d’aménorrhée [SA]) de ces drains doit être minutieuse, car des séquelles à type de déformations de la paroi thoracique ont été décrites.6
À la naissance, la prise en charge dépend de la tolérance respiratoire néonatale. En cas de détresse respiratoire, une évaluation pédiatrique spécialisée et une imagerie thoracique en urgence sont nécessaires. Chez les nouveau-nés asymptomatiques (majorité des cas), un examen clinique pédiatrique est réalisé à la naissance, puis complété à distance par un scanner thoracique ou par une imagerie par résonance magnétique (IRM), afin de confirmer la persistance de la lésion et d’en préciser l’extension. Le suivi est organisé avec les équipes de chirurgie pédiatrique (déjà rencontrées en anténatal). Même lorsqu’elle paraît avoir régressé en anténatal, la malformation peut persister et exposer à un risque d’infections respiratoires récidivantes, de complications compressives ou, plus rarement, de dégénérescence. Une résection chirurgicale programmée peut donc être discutée au cas par cas, en fonction de la taille de la lésion, de sa localisation, de l’existence de symptômes et des pratiques de l’équipe référente.
Séquestration bronchopulmonaire
La séquestration bronchopulmonaire (SBP) est associée à une vascularisation systémique aberrante et communique rarement avec le système bronchique. Il s’agit de tissu pulmonaire entouré de plèvre, non ventilé et alimenté par des vaisseaux artériels d’origine systémique. Le drainage de cette malformation se fait par une veine systémique ou par les veines pulmonaires. Ces lésions sont asymptomatiques à la naissance dans plus de 80 % des cas. Contrairement aux MAKP, les séquestrations sont associées à une autre malformation dans 40 % des cas. La séquestration pulmonaire a une incidence comprise entre 0,15 et 0,18 % des maladies pulmonaires congénitales.7 L’aspect échographique est celui d’une masse hyperéchogène, sans kyste le plus souvent, de contours mieux définis que les MAKP. La visualisation d’un pédicule vasculaire signe le diagnostic de SBP, mais il existe des formes mixtes associant SBP et MAKP. La surveillance à mettre en place est la même que pour les autres maladies pulmonaires. Dans plus de la moitié des cas, ces lésions régressent, mais plus rarement, en cas de séquestre de grande taille avec un débit important dans l’artère qui alimente la masse, il faut se méfier de la survenue d’un possible « effet shunt » avec l’apparition d’une insuffisance cardiaque. Différentes options thérapeutiques ont été décrites pour la prise en charge de ces SBP compliquées, mais l’occlusion du pédicule artériel par embolisation par laser échoguidée semble avoir un réel bénéfice pour les SBP compliquées d’anasarque, avec une survie périnatale proche de 100 % et un taux de complications inférieur à 10 % (prématurité, revascularisation et hématome thoracique).8 Cette procédure échoguidée est réalisée sous anesthésie locale ou locorégionale. Le premier temps de la procédure consiste en la réalisation d’une anesthésie fœtale et en l’administration d’un curare (comme pour la procédure de pose de drain pleuro-amniotique). Le pédicule afférent est ensuite abordé par ponction directe à l’aide d’une aiguille de 17 gauges pour introduire une fibre optique de 400 μm de diamètre permettant de réaliser des tirs de laser avec incrément de puissance de 10 à 40 W. L’efficacité est contrôlée par l’absence de flux en mode Doppler couleur au décours immédiat de la procédure et par la levée de l’anasarque fœtale à distance de la procédure.
L’attitude post-natale est assez similaire à celle adoptée face à une MAKP. Une résection chirurgicale programmée ou une embolisation peuvent être discutées au cas par cas, selon la taille de la lésion, sa vascularisation, son évolution et l’existence de symptômes.
2. Bellini C, Ergaz Z, Boccardo F, et al. Dynamics of pleural fluid effusion and chylothorax in the fetus and newborn: Role of the lymphatic system. Lymphology 2013;46(2):75‑84.
3. Picone O, Benachi A, Mandelbrot L, et al. Thoracoamniotic shunting for fetal pleural effusions with hydrops. Am J Obstet Gynecol 2004;191(6):2047‑50.
4. Lau CT, Kan A, Shek N, et al. Is congenital pulmonary airway malformation really a rare disease? Result of a prospective registry with universal antenatal screening program. Pediatr Surg Int 2017;33(1):105‑8.
5. Adzick NS, Harrison MR, Crombleholme TM, et al. Fetal lung lesions: Management and outcome. Am J Obstet Gynecol 1998;179(4):884‑9.
6. Merchant AM, Peranteau W, Wilson RD, et al. Postnatal chest wall deformities after fetal thoracoamniotic shunting for congenital cystic adenomatoid malformation. Fetal Diagn Ther 2007;22(6):435‑9.
7. Corbett HJ, Humphrey GME. Pulmonary sequestration. Paediatr Respir Rev 2004;5(1):59‑68.
8. Grozdeva L, Senat MV, Vandewynckele N, et al Antenatal management of bronchopulmonary sequestration by intrafetal vascular laser ablation under ultrasound control: Narrative review of the literature and report of three cases. Fetal Diagn Ther 2021;48(1):34‑42.