Les anomalies de fermeture du tube neural représentent les anomalies du système nerveux central les plus fréquentes. Elles peuvent concerner l’étage céphalique (anencéphalie, exencéphalie) ou les vertèbres et la moelle pour les dysraphismes.
Dysraphismes ouverts ou fermés
Seule la prévalence des dysraphismes ouverts (spina bifida aperta) est connue, voisine en France de 5 pour 10 000 grossesses, avec une prévalence pour les naissances vivantes de 1,1 pour 10 000 naissances.1 Le taux de dépistage prénatal pour les dysraphismes ouverts dépasse 95 % en France, avec un taux d’interruption médicale de la grossesse (IMG) voisin de 90 %. Ainsi, environ 120 à 140 enfants naissent en France chaque année avec un dysraphisme ouvert.2
La prévalence des dysraphismes fermés est moins bien connue.
Les causes de survenue pour ce groupe de malformations sont multifactorielles, et plusieurs facteurs de risque sont identifiés : carence en folates, certaines pathologies maternelles antérieures à la grossesse (diabète), obésité maternelle, exposition à certains médicaments tératogènes (notamment antiépileptiques). Les causes génétiques ne sont pas prédominantes en dehors des dysraphismes ouverts associés à plusieurs autres malformations.
Prévention primaire des dysraphismes ouverts encore insuffisante
Les dysraphismes ouverts représentent un groupe de malformations pour lequel il existe une prévention primaire par supplémentation préconceptionnelle en acide folique ; elle est associée à une réduction d’environ 70 % du risque de survenue. La recommandation d’une supplémentation périconceptionnelle (à raison de 0,4 mg/j d’acide folique3 dès quatre à six semaines avant la conception et poursuivie au premier trimestre de la grossesse) existe en France depuis plus de vingt ans, mais son observance est très mauvaise. En effet, selon une étude sur dix ans des données du Système national d’information inter-régimes de l’Assurance maladie (Sniram), ce schéma n’est respecté que pour 15 % des grossesses.4 Cet échec de santé publique explique que la prévalence des dysraphismes ouverts n’a pas diminué dans les vingt dernières années en France. Ce constat devrait inciter à une évolution vers une supplémentation en acide folique de produits alimentaires de base comme les farines.
Différents types de dysraphisme
La moelle épinière se forme durant les premières semaines de développement. Les grandes étapes embryologiques sont la gastrulation (apparition de la chorde, qui induit la différenciation en neuroderme), la neurulation primaire (passage d’une plaque à un tube neural), puis la neurulation jonctionnelle (zone thoracolombaire) et enfin la neurulation secondaire, qui concerne le pôle caudal.
Jusqu’à récemment, les dysraphismes étaient classés selon leur origine embryologique supposée, mais cette vision restait insuffisante pour les différencier dans leur caractère composite.
Quatre facteurs anormaux peuvent concourir aux symptômes neurologiques :
l’ouverture : la fuite de liquide cérébrospinal (LCS) entraîne une malformation et une déformation du pôle céphalique, et peut se compliquer d’une hydrocéphalie ;
la dysplasie : la modification d’une structure anatomique entraîne la perte de la fonction correspondante (syndrome lésionnel) ou des fonctions sous-jacentes (syndrome sous-lésionnel) ;
des facteurs environnementaux : la moelle est normalement entourée de pie-mère au sein d’un sac dural étanche contenant du LCS. En cas de dysraphisme, l’environnement est différent d’un point de vue chimique (exposition au liquide amniotique dans les dysraphismes ouverts), histologique (interface graisse-moelle dans les lipomes) ou bactériologique (infections secondaires en cas de dysraphisme ouvert ou en lien avec un sinus dermique) ;
la sollicitation mécanique : la moelle est normalement reliée à son environnement par des nerfs spinaux, le ligament dentelé et un filum, permettant une mobilité pulsatile. Un stress mécanique imposé par compression ou traction sur la moelle épinière peut provoquer des modifications microvasculaires, et ainsi un défaut de fonctionnement.
Des caractéristiques anatomiques des dysraphismes spinaux correspondent à ces quatre facteurs pourvoyeurs de symptômes et ont mené à une classification en catégories supposées homogènes en pronostic et/ou en prise en charge.
Dysraphismes ouverts
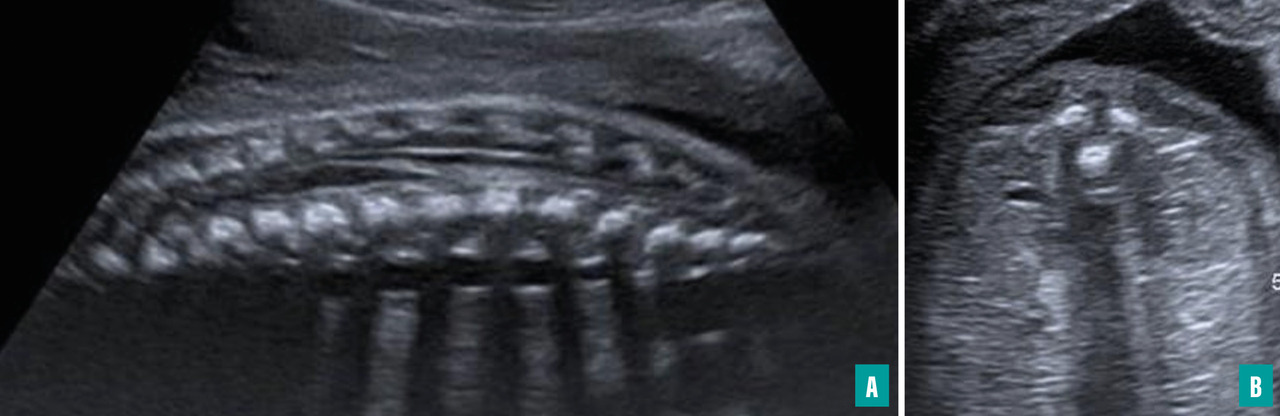
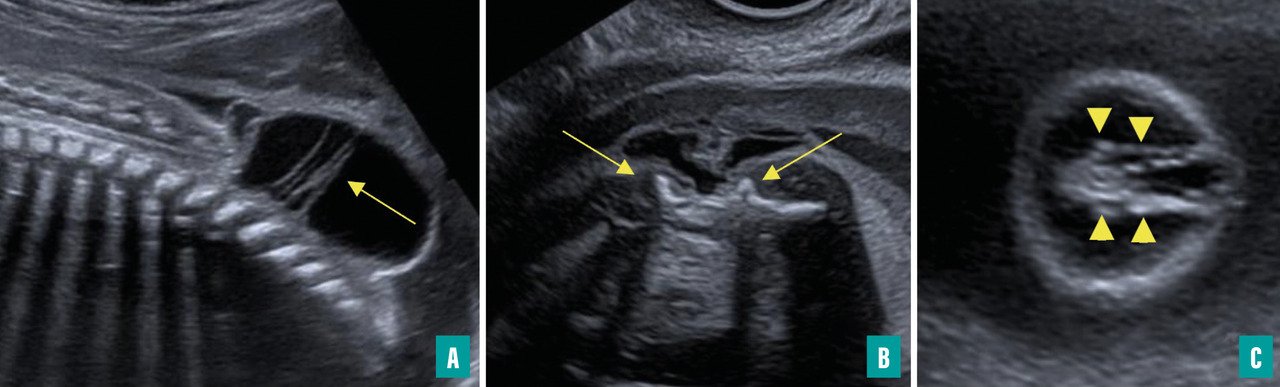
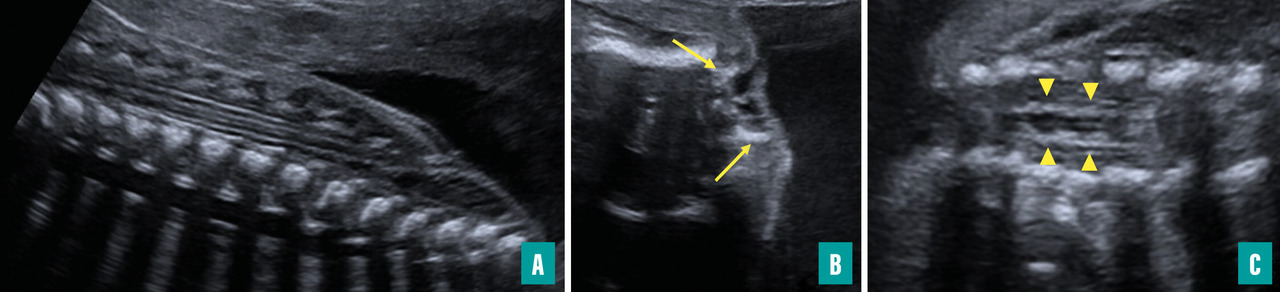
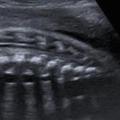
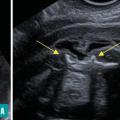
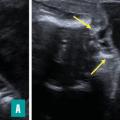
Ils sont représentés principalement par la myéloméningocèle (fig. 1 et 2) et le myéloschisis (fig. 3). Leur retentissement est global sur le système nerveux central et expose donc à des troubles du neurodéveloppement et à un risque de défaut d’autonomie. Une entité intermédiaire avec un moindre retentissement a été décrite et nommée MyeLDM (intermédiaire entre myéloméningocèle et limited dorsal myeloschisis [LDM]).
Moelles épinières dédoublées
Elles sont caractérisées par un cordon médullaire dédoublé au moins à un endroit sur sa longueur, avec possiblement des sacs duraux séparés par un septum osseux (de type I ou diastématomyélie) ou un sac unique avec les deux hémi-moelles (de type II).
Lipomes médullaires
Ils sont caractérisés par la présence anormale de tissu graisseux normal au contact d’une moelle avec effraction de la pie-mère.
Dysraphismes à tige
Ils ont une communication tissulaire linéaire anormale qui attache la partie postérieure de la moelle à la surface cutanée. On leur donne depuis une quinzaine d’années le nom de LDM (limited dorsal myeloschisis) ou de sinus dermique quand le tractus est composé d’épithélium malpighien jusqu’à la dure-mère et expose ainsi à une contamination bactérienne intradurale (méningite, empyème).
Filum fibrolipomateux
Le filum lipomateux est épaissi et rigide et peut entraîner une souffrance médullaire terminale.
Imagerie prénatale : surtout l’échographie
L’analyse des dysraphismes en imagerie prénatale repose essentiellement sur l’échographie, l’imagerie par résonance magnétique (IRM) ayant peu d’indications.5,6
Échographie
L’étude échographique porte essentiellement sur le crâne, le cerveau, le rachis et la moelle ainsi que sur les membres inférieurs. Le principal enjeu est de savoir statuer sur le caractère ouvert ou fermé du dysraphisme. Dans certains cas, la masse des tissus mous rétrorachidiens, dans les formes sacculaires, attire l’attention en premier. Dans d’autres cas (dysraphisme ouvert non sacculaire), les anomalies crâniennes et cérébrales sont vues initialement et conduisent à une étude détaillée du rachis.
Les différents éléments en faveur d’un dysraphisme ouvert ou fermé sont détaillés ci-dessous.6 L’utilisation de sondes en haute fréquence est primordiale pour l’analyse de la moelle et du contenu d’un éventuel sac.
Pôle céphalique
L’aplatissement des os frontaux se voit dans les dysraphismes ouverts et est plus marqué en début qu’en fin de grossesse.
La microcéphalie est un signe fréquemment observé dans les dysraphismes ouverts.
La malformation de Chiari de type II est l’apanage des dysraphismes ouverts et se caractérise par une petite fosse postérieure avec verticalisation de la tente du cervelet, disparition des espaces liquidiens de la fosse postérieure, absence de visibilité des bords des hémisphères cérébelleux et hernie de la partie inférieure du cervelet au travers du foramen magnum. Le quatrième ventricule est abaissé.
La dilatation ventriculaire est fréquente dans les dysraphismes ouverts.
Des hétérotopies sous-épendymaires peuvent être présentes dans les dysraphismes ouverts.
Il existe fréquemment des anomalies du corps calleux dans les dysraphismes ouverts.
Les anomalies de la gyration sont très rarement observées dans les dysraphismes ouverts et ne sont pas constatées dans les dysraphismes fermés.
Rachis
En regard du défect rachidien, la direction des lames est étudiée sur des coupes axiales : la convergence des lames se voit dans les dysraphismes fermés, leur divergence avec éversion ou leur parallélisme sont l’apanage des formes ouvertes.
Dans certains cas de dysraphisme ouvert, il peut y avoir une cyphose, généralement lombaire.
Moelle
Dans les dysraphismes ouverts, la hernie du cervelet dans le foramen magnum peut apparaître comme une structure hyperéchogène en arrière de la moelle cervicale.
Le trajet de la moelle peut être anormal : attraction postérieure à n’importe quel niveau dans les LDM (avec, dans les formes sacculaires, une partie du trajet dans le sac, puis un retour dans le canal rachidien), moelle double dans les diastématomyélies (importance d’analyser le diamètre des deux moelles en coupes axiales).
La moelle terminale peut avoir un aspect anormal : perte du renflement physiologique, aspect effilé dans une moelle bas attachée, aspect tronqué dans un syndrome de régression caudale, masse hyperéchogène au contact dans un lipome, terminaison par une placode dans un dysraphisme ouvert.
La moelle terminale peut être en position anormale : basse ou, au contraire, trop haute (régression caudale), au sein d’une méningocèle.
Tissus mous prérachidiens
La présence d’un sac est possible, avec des caractéristiques variables : siège, taille, paroi (épaisse [dysrasphisme fermé], en continuité avec les téguments adjacents ou fine [dysraphisme ouvert]), contenu (présence ou non de tige(s) fibroneurale(s) [LDM], de racines nerveuses [dysrasphisme ouvert], de la moelle [dysrasphisme fermé], d’une placode neurale au dos du sac [dysrasphisme ouvert]).
Un lipome sous-cutané peut aussi s’observer.
Membres inférieurs
Il peut exister une malposition des pieds, une amyotrophie des membres inférieurs dans les dysraphismes ouverts.
Une anomalie de la mobilité des membres inférieurs est également possible.
Indications de l’IRM
L’IRM peut être indiquée en cas de mauvaises conditions échographiques (liées à la paroi maternelle et à la position dos postérieur) et de suspicion de dysraphisme lors de l’analyse du pôle céphalique.
L’IRM est systématiquement réalisée à titre pré- et postopératoire quand une intervention in utero est envisagée.
Elle permet en général de mieux analyser qu’à l’échographie certains éléments du pôle céphalique : le vermis, les espaces liquidiens de la fosse postérieure (lorsqu’ils sont quasi virtuels), les hétérotopies sous-épendymaires.
Toutefois, en règle générale, si l’analyse échographique de la moelle est réalisée avec une sonde de haute fréquence, elle est toujours plus précise que celle de l’IRM.
Prise en charge prénatale
La prise en charge prénatale dépend de la caractérisation du type de dysraphisme. Il est indispensable que cette prise en charge repose sur une équipe multidisciplinaire impliquant les spécialistes du diagnostic prénatal, les radiopédiatres, les neurochirurgiens et parfois des spécialistes de médecine physique et de réadaptation de l’enfant.
Dysraphismes ouverts
Pour les dysraphismes ouverts, un geste ovulaire invasif est souvent proposé pour une étude du caryotype et une analyse chromosomique sur puce à ADN (Acpa), même si les dysraphismes ouverts isolés sont très rarement associés à une anomalie cytogénétique. La place de l’exome ou d’une étude du génome reste à préciser.
Compte tenu de la sévérité du pronostic, associant une paralysie des membres inférieurs (dont le tableau dépend de la hauteur du défect) à une incontinence sphinctérienne urinaire (vessie neurologique) et fécale, une IMG est recevable. Cependant, la possibilité de réaliser une chirurgie fœtale de réparation, qui est accessible en France depuis 2014, doit désormais faire partie de l’information pronostique en prénatal.7 Celle-ci n’efface, bien sûr, pas les handicaps mais a pour but de « protéger » le cerveau fœtal (en stoppant la fuite de LCS et en permettant ainsi de corriger ou de limiter la malformation de Chiari de type II), donc de réduire la ventriculomégalie secondaire et la nécessité de recourir à une dérivation ventriculo-péritonéale post-natale et enfin d’améliorer le pronostic moteur.
Deux types de réparation
Il existe actuellement deux types de réparation prénatale. La première, développée depuis plus de vingt ans, correspond à la chirurgie fœtale de réparation dite « à ciel ouvert ».8 Actuellement, cette intervention est réservée aux dysraphismes ouverts (myéloméningocèles et myéloschisis) de S1 à T1, strictement isolés, et dans le cadre d’une grossesse singleton.
Les contre-indications sont strictes et incluent une anomalie fœtale associée (et notamment une angulation anormale du rachis fœtal), une contre-indication maternelle (pathologie infectieuse chronique, hypertension artérielle, diabète ou indice de masse corporelle [IMC] supérieur à 40 kg/m2), un surrisque a priori d’accouchement prématuré (col court, antécédent d’accouchement prématuré).
Cette intervention doit être réalisée entre 23 et 26 semaines d’aménorrhée (SA). Elle se déroule sous anesthésie générale maternelle, après mise en place au préalable d’un cathéter de péridurale, qui participe, en postopératoire, à la fois à l’analgésie maternelle et au bon relâchement utérin. L’opération débute par une laparotomie de type Pfannenstiel permettant l’extériorisation de l’utérus gravide. Après repérage du la position du placenta, une hystérotomie est réalisée sur quelques centimètres (avec sutures des berges pour assurer l’hémostase et fixer les membranes) permettant d’avoir accès à la cavité amniotique.9 Le fœtus est ensuite positionné pour permettre d’exposer la malformation (il est maintenu en permanence dans l’utérus). Après complément d’anesthésie fœtale par injection intramusculaire, une dissection des contours de la placode neurale est ensuite réalisée. Puis la moelle est recouverte par un premier plan de fermeture à partir des vestiges de la dure-mère. Un deuxième plan musculaire (muscles paravertébraux) et enfin une fermeture cutanée sont réalisés, permettant d’obtenir une réparation « étanche » et ainsi stopper la fuite de LCS. Une hystéroraphie en deux plans et une fermeture plan par plan terminent le geste.
Les bénéfices de cette chirurgie ont été démontrés par un essai randomisé, avec en moyenne un gain d’un à deux corps vertébraux sur le plan lésionnel (et donc un bénéfice moteur) et une réduction de moitié de la nécessité de recourir à une dérivation ventriculo-péritonéale en post-natal en comparaison à un groupe de fœtus avec réparation post-natale.8 Les bénéfices sur l’incontinence urinaire sont plus discutés, mais le devenir urologique pourrait être meilleur pour les enfants d’âge scolaire.
Risques maternels et fœtaux
Cette chirurgie comporte des risques, qui doivent être clairement exposés. Les risques maternels périopératoires sont dominés par un risque d’hémorragie, d’hématome rétroplacentaire (6 %), d’œdème aigu pulmonaire (6 %) – même si ce risque semble très inférieur dans notre expérience du fait de la modification des protocoles d’anesthésie maternelle et de tocolyse. La morbidité obstétricale doit également être clairement expliquée, avec, du fait de l’hystérotomie corporéale, la mise en évidence d’une déhiscence au niveau de la cicatrice à la naissance dans 3 à 10 % des cas et la nécessité de réaliser une césarienne pour la grossesse en cours et les grossesses suivantes. Il existe par ailleurs un risque de rupture utérine pour les grossesses suivantes, qui est évaluée entre 3 à 10 % et nécessite une surveillance particulière.10 La principale complication pour le fœtus opéré est représentée par un risque d’accouchement prématuré et de rupture prématurée des membranes observée dans 46 % des cas. Ainsi, si le terme médian de naissance rapporté dans les études est voisin de 34 SA, environ 13 % des naissances surviennent avant 30 SA.8,9
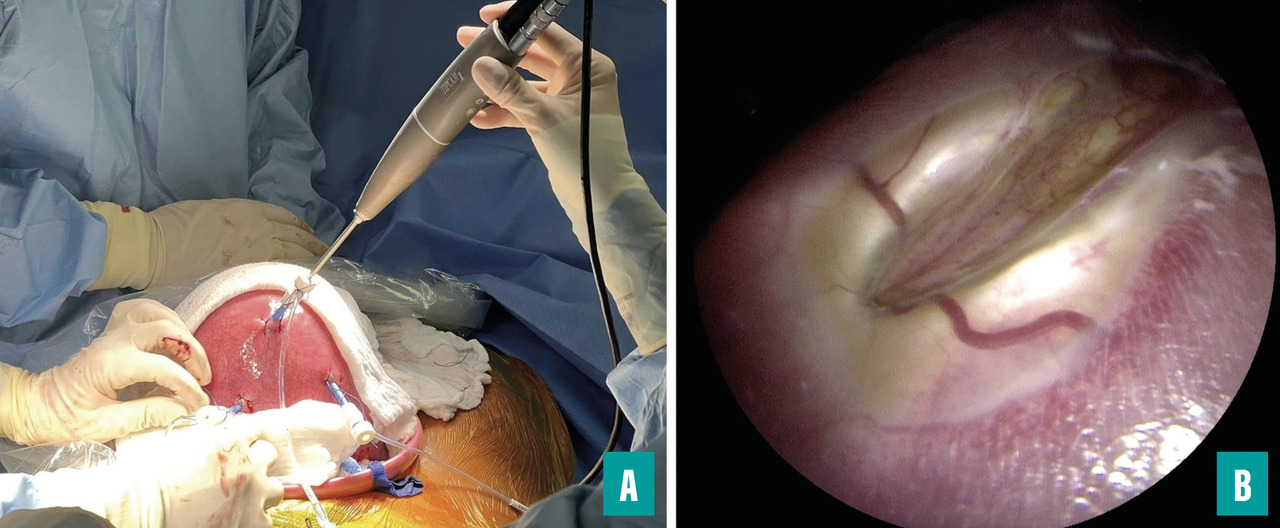
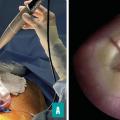
Réparation fœtoscopique
Du fait de la morbidité obstétricale associée à la chirurgie à ciel ouvert, des techniques de réparation fœtoscopique ont été développées.9,10 Globalement, deux approches sont utilisées dans le monde : la réparation fœtoscopique transcutanée avec trois trocarts ou la réparation fœtoscopique laparo-assistée, pour laquelle l’utérus est d’abord exposé après laparotomie et ensuite deux à trois trocarts sont introduits. Cette dernière technique est celle proposée à l’hôpital Trousseau et utilisée par la majorité des centres européens, notamment du fait du taux de rupture des membranes bien supérieur (supérieur à 60 %) dans l’approche purement transcutanée (fig. 4).11,12 Les développements de la fœtoscopie devraient permettre d’obtenir une qualité de réparation équivalente à celle de la réparation à ciel ouvert (actuellement, le taux de reprise de la cicatrice après la naissance est voisin de 10 %) et de réduire le risque d’accouchement prématuré et de rupture des membranes, qui est actuellement supérieur à celui observé en cas de réparation à ciel ouvert.13 Cependant, l’avantage d’ores et déjà indiscutable de la réparation fœtoscopique est représenté par le fait que les orifices de trocarts ne sont pas associés à un risque de déhiscence utérine, permettant un accouchement par les voies naturelles sans risque de rupture utérine pour les grossesses ultérieures.14
Trois options possibles
La prise en charge prénatale des dysraphismes ouverts a ainsi connu de profonds bouleversements dans les deux dernières décennies. Des développements supplémentaires (avec notamment l’utilisation de thérapies complémentaires à la chirurgie fœtale telles que la thérapie cellulaire) sont encore attendus.16 Néanmoins, avec un taux de dépistage de ces anomalies supérieur à 90 % en France, une majorité des patientes optent pour la réalisation d’une IMG. L’information prénatale doit ainsi être complète, loyale et éclairée sur les trois options possibles : naissance et prise en charge post-natale, chirurgie fœtale ou IMG.16
Dysraphismes fermés
Les dysraphismes fermés, notamment suspectés devant la découverte d’une image anormale sacculaire en regard du rachis (souvent en région dorsolombaire, sans anomalie cérébrale associée), peuvent être plus difficiles à caractériser en prénatal.16 La prise d’’avis auprès d’un centre expert doit être favorisée afin d’offrir aux patientes une information loyale et éclairée (centre de référence maladies rares spin@ [CRMR spin@] : https ://crmr-spina.fr). Le plus souvent, le pronostic moteur est meilleur, voire normal. La possibilité d’un tableau de vessie neurologique avec risque d’incontinence doit être évaluée au cas par cas, en prenant en compte le niveau du défect et le type de dysraphisme.17,18 La chirurgie des dysraphismes fermés ne fait pas toujours l’objet de consensus et encore moins d’une justification scientifique de haut niveau de preuve, sa décision est donc prise au cas par cas.
Prise en charge post-natale
La prise en charge post-natale est adaptée aux caractéristiques du dysraphisme et à la symptomatologie. La règle est une prise en charge pluridisciplinaire au long cours : médecine physique et rééducation, urologie, orthopédie, neurochirurgie, accompagnements psychologique et social.
La chirurgie des dysraphismes ouverts est systématique, consistant en une fermeture néonatale (quand elle n’a pas été réalisée en anténatal) précoce (dans les quarante-huit heures) avec libération de l’attache médullaire terminale et restauration des plans anatomiques duraux, aponévrotiques et cutanés. En cas de nécessité, un geste de dérivation du LCS peut être associé, classiquement durant la première année de vie.
Le grand principe de la chirurgie d’un dysraphisme est d’optimiser l’environnement mécanique, histologique et chimique d’une moelle potentiellement dysplasique (et qui le restera). La décision opératoire est sous-tendue par l’amélioration de l’histoire naturelle de la malformation mise en balance avec un risque d’aggravation neurologique iatrogénique. On parle de geste de libération médullaire.
Suivi et devenir
Pour tous les types de dysraphisme (qu’il y ait eu une intervention neurochirurgicale ou non), un suivi à l’âge pédiatrique, au moment de l’âge de transition et au cours de la vie adulte est indispensable. Ce suivi est le plus souvent multidisciplinaire et implique neurochirurgiens, spécialistes de médecine physique et de réadaptation, orthopédistes et souvent urologues. Une attention particulière doit être portée sur l’accompagnement à la sexualité et l’anticipation du suivi de la grossesse chez les femmes. Ce suivi peut être aidé grâce au maillage territorial représenté par les centres de référence maladies rares dédiés (CRMR spin@).
2. Peyronnet B, Gao F, Brochard C, et al. Épidémiologie du spina bifida en France. Prog Urol 2016;26:721.
3. Organisation mondiale de la santé, Supplémentation périconceptionnelle en folates avec ou sans multivitamines dans la prévention des anomalies du tube neural.
4. de la Fourniere B, Dhombres F, Maurice M, et al. Prevention of neural tube defects by folic acid supplementation: A national population-based study. Nutrients 2020;12(10):3170.
5. Guilbaud L, Carreras E, Garel C, et al. Workgroup Spina Bifida and other Dysraphisms (SBoD), ITHACA‐eUROGEN European reference networks. Proposal for standardized prenatal assessment of fetal open dysraphisms by the European reference network for Intellectual disability, TeleHealth, Autism and Congenital Anomalies (ITHACA) and eUROGEN. Prenat Diagn 2024;44(9):1073-87.
6. Vande Perre S, Guilbaud L, de Saint-Denis T, et al. The myelic limited dorsal malformation: Prenatal ultrasonographic characteristics of an intermediate form of dysraphism. Fetal Diagn Ther 2021;48(9):690-700.
7. Jouannic JM, Guilbaud L, Maurice P, et al. The ethics of fetal myelomeningocele surgery. Gynecol Obstet Fertil Senol 2022;50(2):189-93.
8. Adzick NS, Thom EA, Spong CY, et al. MOMS Investigators. A randomized trial of prenatal versus postnatal repair of myelomeningocele. N Engl J Med 2011;364(11):993-1004.
9. Guilbaud L, Maurice P, Lallemant P, et al. Open fetal surgery for myelomeningocele repair in France. J Gynecol Obstet Hum Reprod 2021;50(9):102155.
10. Goodnight WH, Bahtiyar O, Bennett KA, et al.; fMMC Consortium sponsored by NAFTNet. Subsequent pregnancy outcomes after open maternal-fetal surgery for myelomeningocele. Am J Obstet Gynecol 2019;220(5):494.e1-494.e7.
11. Giné C, Arévalo S, Maíz N, et al. Fetoscopic two-layer closure of open neural tube defects. Ultrasound Obstet Gynecol 2018;52(4):452-7.
12. Sanz Cortes M, Chmait RH, Lapa DA, et al. Experience of 300 cases of prenatal fetoscopic open spina bifida repair: Report of the International Fetoscopic Neural Tube Defect Repair Consortium. Am J Obstet Gynecol 2021;225(6):678.e1-678.e11.
13. Belfort MA, Whitehead WE, Shamshirsaz AA, et al. Fetoscopic open neural tube defect repair: Development and refinement of a two-port, carbon dioxide insufflation technique. Obstet Gynecol 2017;129(4):734-43.
14. Sanz Cortes M, Shamshirsaz AA, Ugoji CH, et al. Outcomes of subsequent pregnancies after 2-port laparotomy-assisted fetoscopic spina bifida repair. Am J Obstet Gynecol 2021;225(4):452-4.
15. Athiel Y, Jouannic JM, Mauffre V, et al. Allogenic umbilical cord-derived mesenchymal stromal cells improve motor function in prenatal surgical repair of myelomeningocele: An ovine model study. BJOG 2024;131(6):759-67.
16. Jouannic JM, Guilbaud L, Maurice P, et al. The ethics of fetal myelomeningocele surgery. Gynecol Obstet Fertil Senol 2022;50(2):189-93.
17. Friszer S, Dhombres F, Morel B, et al. Limited dorsal myeloschisis: A diagnostic pitfall in the prenatal ultrasound of fetal dysraphism. Fetal Diagn Ther 2017;41(2):136-44.
18. Pang D, Zovickian J, Oviedo A, et al. Limited dorsal myeloschisis: A distinctive clinicopathological entity. Neurosurgery 2010;67(6):1555-79.