La hernie de coupole diaphragmatique (HCD) est une malformation rare dont la fréquence est d’environ 1/3 000naissances.1 Elle se caractérise par un défaut de fermeture du diaphragme ayant pour conséquence l’ascension dans le thorax de viscères abdominaux comme l’estomac, le tube digestif et/ou le foie. Le développement pulmonaire, pendant la vie fœtale, en est altéré à des degrés variables, et les nouveau-nés présentent une hypoplasie pulmonaire, le plus souvent associée à une hypertension artérielle pulmonaire.
Depuis les premières interventions de chirurgie in utero à utérus ouvert au début des années 1990, la prise en charge des fœtus atteints de HCD a beaucoup évolué. Son taux de dépistage, en France, est proche de 90 %.1 En 2008, la création du centre de référence maladies rares (CRMR) rattaché à la filière de santé maladies rares FIMATHO (Filière des maladies rares abdomino-thoraciques) a permis de mettre en place un maillage territorial assurant une expertise relativement homogène sur le territoire. Un protocole national de diagnostic et de soins (PNDS) a été mis à jour en 2020 ;2 il permet d’orienter les équipes pluridisciplinaires prenant en charge les enfants atteints d’une HCD, de la vie fœtale à l’âge adulte. Des recommandations européennes (ERNICA) ont également été publiées pour harmoniser le suivi prénatal.3
Depuis 2021 et après une dizaine d’années d’évaluation, la chirurgie in utero par occlusion de la trachée fœtale fait partie des options thérapeutiques pour les fœtus porteurs de HCD de formes sévère et modérée.
Évaluation prénatale d’un fœtus porteur d’une HCD
Lorsqu’une HCD est suspectée chez un fœtus, la patiente doit être adressée à un centre pluridisciplinaire de diagnostic prénatal (CPDPN), qui confirme le diagnostic et évalue le pronostic de la malformation. Le diagnostic est porté uniquement à l’échographie, le plus souvent au deuxième trimestre, mais parfois au premier ou au troisième trimestre. L’évaluation du pronostic est le plus souvent effectuée au moment de la confirmation diagnostique, après l’échographie du deuxième trimestre de grossesse.
Pronostic assombri en cas de polymalformations
Le pronostic dépend, en premier lieu, du caractère isolé ou non de la HCD. Tout type d’anomalie peut être associé à une HCD ; les plus fréquentes sont les anomalies cardiaques. Celles-ci sont à différencier de l’hypotrophie, souvent présent dans les formes sévères de HCD gauche. L’existence d’un syndrome polymalformatif grève le pronostic et rend le fœtus non éligible à une thérapie in utero, pour des raisons éthiques. De nombreuses associations à des anomalies génétiques (ou à des syndromes, plus de 70) ont été rapportées, mais aucune d’entre elles n’est spécifiquement liée à la HCD.4
Une amniocentèse avec analyse chromosomique sur puce à ADN (Acpa) est proposée systématiquement. Une anomalie chromosomique peut être mise en évidence dans environ 30 % des cas, en particulier en cas d’association malformative. Un exome fœtal est de plus en plus souvent réalisé mais ne semble pas apporter d’avantage majeur par rapport à l’Acpa pour les formes isolées, en l’état actuel des connaissances.
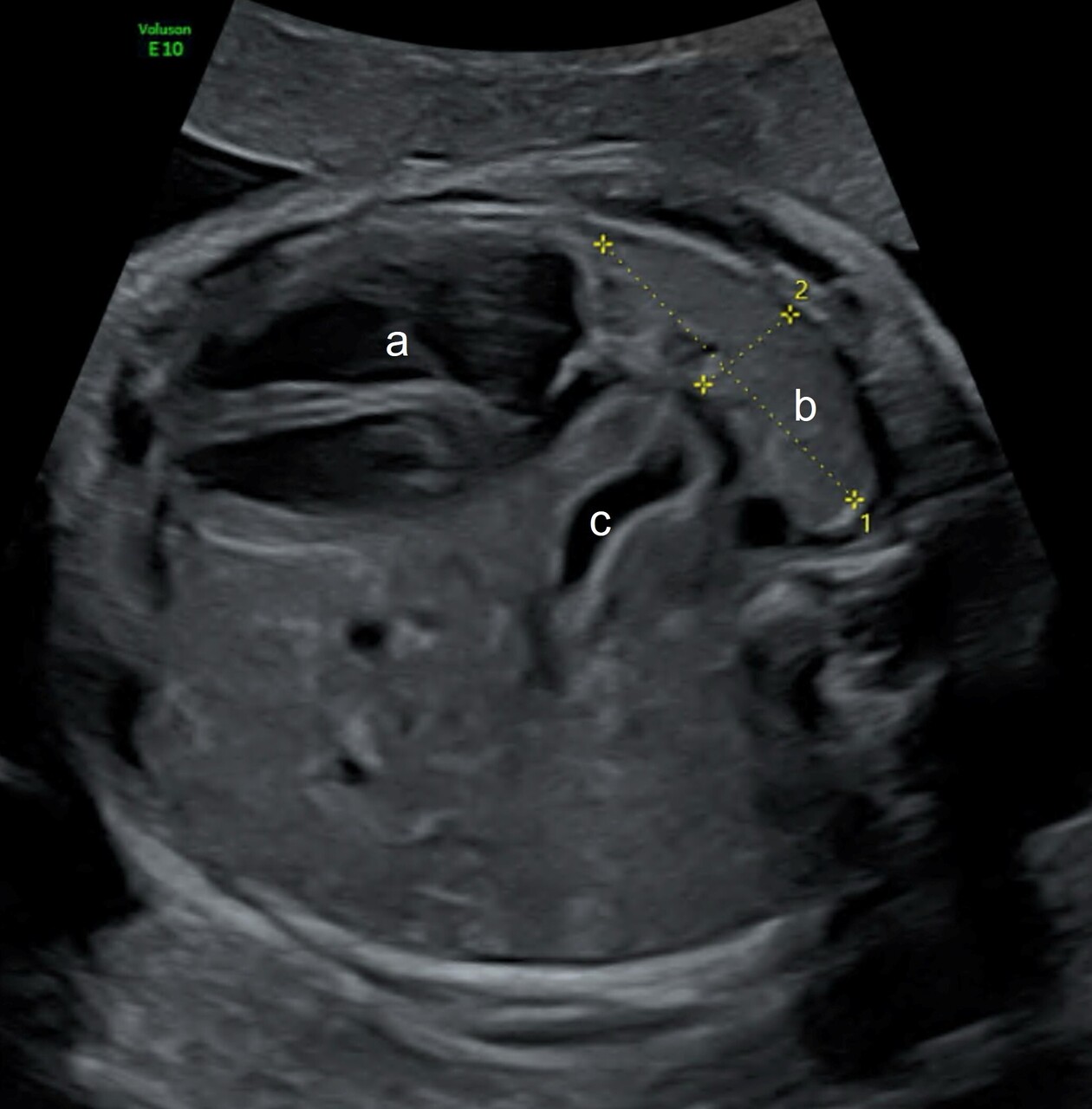
L’évaluation pronostique repose sur plusieurs éléments : la mesure échographique du « lung over head ratio » observé/attendu (LHR o/a) [fig. 1] et l’estimation du volume pulmonaire total o/a (VPT o/a) à l’imagerie par résonance magnétique (IRM). La position de l’estomac permet également de préciser de façon fiable la position du foie (pour les hernies gauches). Celle-ci est en effet déterminée, à l’échographie, en étudiant la position de l’estomac par rapport au plan des valves cardiaques, dans une coupe dite « des quatre cavités » ; plus l’estomac est éloigné de la pointe du cœur, plus le foie est ascensionné. Une IRM est également réalisée pour mesurer le volume pulmonaire total. La bonne mesure du LHR o/a, par des équipes entraînées, est utilisée seule pour évaluer le pronostic et proposer les différents parcours de soins. Le pronostic des HCD diagnostiquées au premier trimestre de la grossesse est moins bon, mais il est recommandé d’attendre l’échographie du cinquième mois pour évaluer le pronostic de façon fiable.5
La HCD est localisée à droite dans environ 20 % des cas. Le pronostic plus défavorable de cette forme de HCD est débattu, mais une HCD droite est considérée de mauvais pronostic si le LHR est inférieur ou égal à 50 %.
Choix de la prise en charge
L’évaluation pronostique, qui établit la sévérité de la HCD, est déterminante pour choisir la prise en charge prénatale la plus adaptée : surveillance attentiste jusqu’à l’accouchement ou chirurgie in utero. Dans tous les cas, l’accouchement doit être réalisé dans une maternité de type III, avec une réanimation néonatale et un service de chirurgie pédiatrique sur site.
Une expertise de la HCD est indispensable à une bonne prise en charge de cette pathologie complexe. En effet, les résultats en matière de survie sont bien meilleurs dans les centres accueillant plus de dix nouveau-nés avec HCD par an.6
En cas de syndrome polymalformatif ou dans certaines formes isolées de très mauvais pronostic, une interruption médicale de grossesse peut être discutée, si elle est demandée par la patiente. Dans cette situation, une information éclairée, par un praticien connaissant bien la pathologie et le devenir des nouveau-nés qui en sont porteurs, est nécessaire. Il est indispensable, dans tous les cas, d’organiser pour la patiente, et l’autre parent, une consultation avec un réanimateur et/ou un chirurgien pédiatre pour apporter une information précise sur la prise en charge de l’enfant à la naissance ainsi qu’en réanimation. Cette consultation permet également d’aborder le devenir à court, moyen et long termes des enfants porteurs de HCD. L’évolution dépend de la sévérité de la forme de HCD. Certains nouveau-nés sont opérés rapidement et sortent du service de réanimation après quelques jours. D’autres restent plusieurs mois en réanimation. Les enfants qui ne sont pas opérés sont les plus gravement atteints et ils décèdent avant de pouvoir l’être.
Idéalement, cette consultation a lieu une fois que l’évaluation pronostique est complète, mais, pour certains couples, il peut être discuté, au cas par cas, d’organiser cet échange plus tôt. Un discours concerté et homogène entre les intervenants de la prise en charge pré- et post-natale est nécessaire pour accompagner au mieux le couple dans sa prise de décision.
Chirurgie in utero
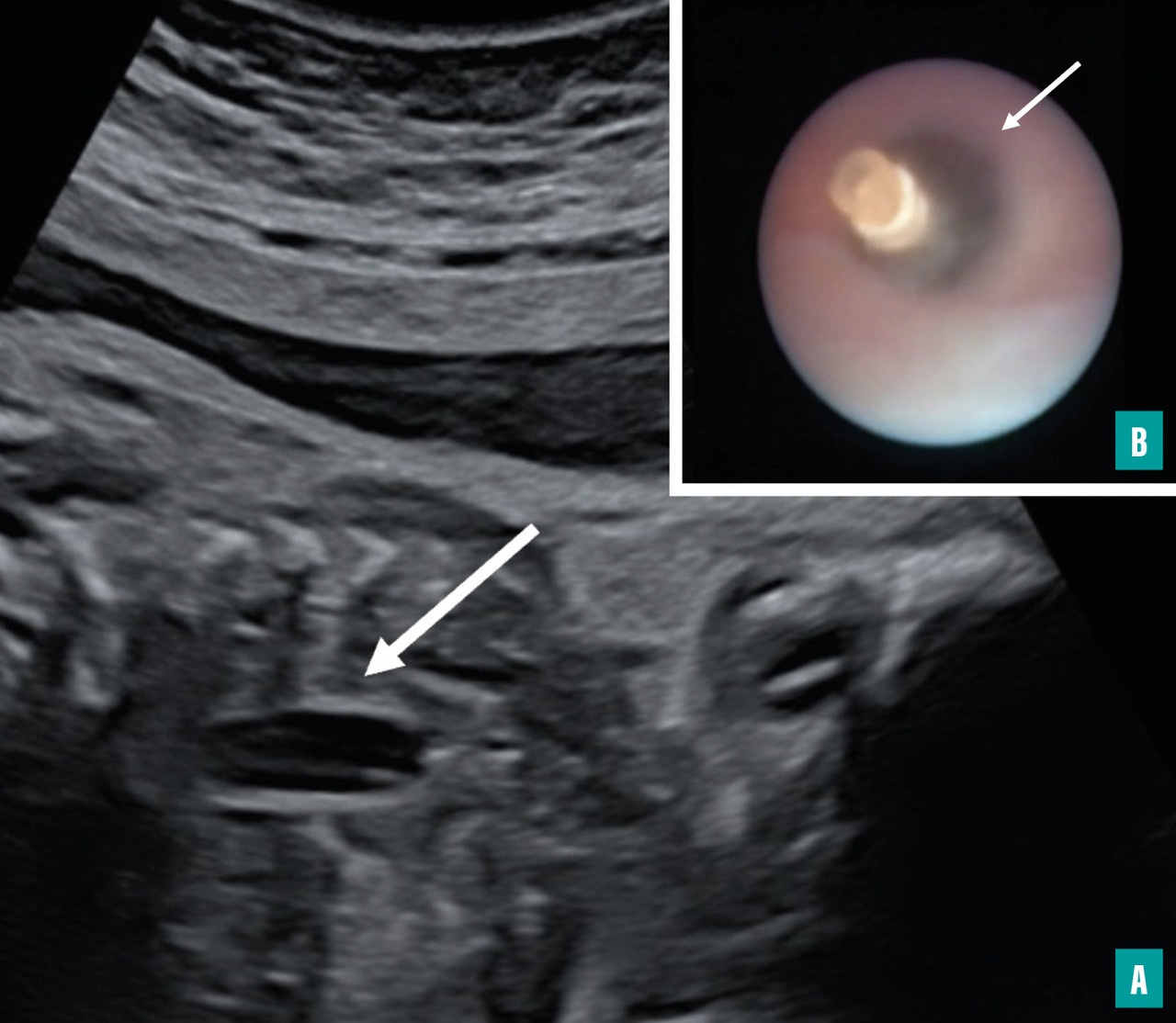
Près de trente ans après le premier article scientifique démontrant, chez le fœtus de brebis porteur d’une HCD, la possibilité d’augmenter le volume pulmonaire en réalisant une occlusion trachéale par fœtoscopie (FETO)* à l’aide d’un ballonnet,7 cette technique a été validée chez l’humain par deux essais randomisés, les essais TOTAL.8,9
Le principe de cette technique repose sur le fait que le poumon fœtal se développe grâce à la présence de liquide synthétisé par les pneumocytes de type 2 et maintenu à une certaine pression dans les futures voies aériennes. Les mouvements respiratoires fœtaux permettent l’ouverture régulière des cordes vocales et l’expulsion de ce liquide dans le liquide amniotique. Cette ouverture dépend, entre autres, du différentiel de pression entre la cavité amniotique et les voies aériennes. En maintenant le liquide dans les poumons fœtaux de façon transitoire, grâce à un ballonnet, le poumon hypoplasique des fœtus porteurs de HCD se développe.
Actuellement, cette technique FETO peut être proposée pour les fœtus ayant une HCD gauche de pronostic sévère ou modéré, c’est-à-dire avec un LHR o/a inférieur à 35 % quelle que soit la position du foie, ou un LHR o/a compris entre 35 et 44,9 % avec un foie en position intrathoracique.
Pour les HCD droites, la fœtoscopie est également proposée pour un LHR inférieur à 50 %.10
Dans les années 1990, la chirurgie in utero était réalisée à utérus ouvert avec pour objectif principal de réintégrer les viscères dans l’abdomen et de lever ainsi la compression pulmonaire. En 1997, un essai randomisé a mis fin à cette technique : il a mis en évidence que les enfants avec les formes les plus sévères de HCD avec foie intrathoracique décédaient à la suite de sa réintégration par plicature du ductus venosus. Les fœtus éligibles étaient donc ceux présentant une forme de meilleur pronostic, et l’essai a démontré qu’il n’y avait pas de bénéfice pour ces fœtus à réaliser une chirurgie à utérus ouvert en matière de survie, de durée d’hospitalisation et d’utilisation de l’Ecmo (« extracorporeal membrane oxygenation », oxygénation par membrane extracorporelle).11 De plus, la morbidité maternelle était loin d’être négligeable. Plusieurs équipes ont alors développé le modèle d’occlusion trachéale par fœtoscopie. Elles ont montré que la période optimale d’occlusion chez le fœtus ovin correspondait, chez l’homme, à la période entre 27 et 32 semaines d’aménorrhée (SA). De plus, l’occlusion devait être réversible car elle entraînait un déficit en surfactant par dédifférenciation des pneumocytes de type 2 en pneumocytes de type 1 (par augmentation de la tension sur la paroi des alvéoles).12
En 2001, Jan Deprest et Kypros Nicolaides ont réalisé la première pose de ballonnet chez l’homme, utilisant le même que celui de l’étude princeps de 1997. Par la suite, les résultats d’un premier essai (TOTAL « formes sévères ») publiés en 2009, sur environ 200 cas de HCD de mauvais pronostic, ont montré un bénéfice en matière de survie.13 Ces résultats encourageants ont permis la mise au point d’un essai randomisé pour les formes gauches isolées modérées, définies par un LHR o/a compris entre 25 et 34,9 % quelle que soit la position du foie, et entre 35 et 44,9 % avec un foie intrathoracique ; ce second essai (TOTAL « formes modérées ») a débuté en 2008 et a été clôturé en 2019. Les deux essais étaient ouverts, randomisés, multicentriques, de supériorité et en groupes parallèles. Le critère de jugement principal était la survie à la sortie de réanimation, et cinq études intermédiaires étaient prévues.
Dans l’essai TOTAL « formes sévères », 95 patientes ont été randomisées, et pour 39 d’entre elles (analyse per protocole), le ballonnet a été posé entre 27 et 29+ 6 SA et retiré par une deuxième fœtoscopie entre 34 et 34+ 6 SA. Cet essai a montré une amélioration statistiquement significative de la survie, qui était de 15 % sans FETO et de 40 % avec FETO.
Dans l’essai TOTAL « formes modérées », 196 patientes ont été randomisées, et pour 88 d’entre elles (analyse per protocole), le ballonnet était posé entre 30 et 30+ 6 SA et retiré par une deuxième fœtoscopie entre 34 et 34+ 6 SA. Le choix d’un terme plus tardif a été fait en raison du risque de prématurité induit par toute manipulation in utero. Cette étude a montré que l’augmentation de la survie de 13 % entre les deux groupes n’était pas significative. Une publication secondaire aux essais14 a montré récemment que si le ballonnet avait été posé plus tôt dans les formes modérées, le taux de survie aurait été meilleur, mais au prix d’un taux de prématurité augmenté. La FETO augmente donc le risque d’accouchement prématuré avec, pour chaque semaine d’insertion plus précoce du ballonnet, un risque associé d’accouchement plus précoce de quatre jours mais un taux de survie augmenté.
La rupture prématurée des membranes (RPM) est le principal effet indésirable de la technique FETO. Dans l’essai « formes sévères », le taux de RPM était de 48 % dans le groupe avec FETO et de 11 % dans le groupe sans FETO. Dans l’essai « formes modérées », il était de 44 % versus 12 %. L’âge gestationnel médian à l’accouchement était de 34,6 SA versus 38,4 SA dans l’essai « formes sévères » et de 35,9 SA versus 38,1 SA dans l’essai « formes modérées ». Il faut souligner que le risque de RPM est plus élevé dans les groupes contrôles des deux études que dans la population générale, ce qui signifie que le simple fait que le fœtus soit porteur d’une HCD augmente le risque d’accoucher avant 37 SA.
Lors de ces deux essais, très peu de complications maternelles ont été rapportées, ce qui confirme les données de la littérature, avec un risque de 3,44 % toutes complications confondues.
Le risque rapporté de RPM et de prématurité est sans doute lié, au moins en partie, à la répétition de la fœtoscopie, geste invasif nécessaire pour la mise en place du ballonnet, puis pour son retrait.
En 2020, un nouveau ballonnet a été mis au point, le ballonnet SMART,15 qui se dégonfle en présence d’un champ magnétique (1,5 tesla). Le ballonnet est semblable à celui utilisé lors des essais TOTAL, mais il est fermé par une bille métallique et un aimant. Le retrait du ballonnet ne nécessite donc pas de réitérer la fœtoscopie. La patiente dont le fœtus a bénéficié d’une pose du ballonnet SMART passe dans le champ magnétique d’une machine d’IRM ; celui-ci permet au ballonnet de se dégonfler en déplaçant la bille métallique. Ce ballonnet a fait l’objet d’un essai de phase I, réalisé par notre équipe et celle de Jan Deprest à Louvain. L’Agence nationale de sécurité du médicament et des produits de santé (ANSM) a autorisé l’équipe du CPDPN Béclère-Bicêtre à l’utiliser à titre compassionnel en attendant son marquage CE.
Devenir et suivi des nouveau-nés traités
Le devenir des enfants porteurs d’une HCD dépend de la sévérité de la forme de HCD. Certains sont opérés rapidement et sortent de réanimation après quelques jours. D’autres restent plusieurs mois en réanimation.
Les enfants qui ne sont pas opérés ont les formes les plus graves et ils décèdent avant de pouvoir l’être. Le devenir à long terme des enfants est bon, mais certains suivent un parcours médical parfois long, avec des complications respiratoires, des troubles de l’oralité et parfois une scoliose. Un suivi régulier dans le cadre d’un centre de référence est recommandé.
Le suivi à long terme des enfants ayant bénéficié d’une FETO est évidemment nécessaire et est en cours d’évaluation.
La chirurgie améliore la survie
La technique FETO améliore le pronostic des enfants porteurs de HCD. Le terme optimal pour la pose du ballonnet semble être compris entre 27 et 29 + 6 SA, et le retrait, idéalement non invasif, vers 34 SA. Cette technique améliore significativement le taux de survie et n’est, à ce jour, proposée qu’aux fœtus ayant une HCD de pronostic sévère ou modéré. La réanimation de ces nouveau-nés nécessite une bonne connaissance des particularités de leur prise en charge. Une attention particulière doit être portée à l’égalité d’accès à ces soins très spécifiques sur l’ensemble du territoire français.
2. Haute Autorité de santé. Hernie de coupole diaphragmatique. Protocole national de diagnostic et de soins, 2020.
3. Russo FM, Cordier AG, De Catte L, et al. Proposal for standardized prenatal ultrasound assessment of the fetus with congenital diaphragmatic hernia by the European Reference Network on rare Inherited and Congenital Anomalies (ERNICA). Prenat Diagn 2018;38:629-37.
4. Zani A, Chung WK, Deprest J, et al. Congenital diaphragmatic hernia. Nat Rev Dis Primers 2022;8:37.
5. Bouchghoul H, Senat MV, Storme L, et al. Center for rare diseases for congenital diaphragmatic hernia. Congenital diaphragmatic hernia: Does gestational age at diagnosis matter when evaluating morbidity and mortality? Am J Obstet Gynecol 2015;213:535.e1-7.
6. Mehl SC, Portuondo JI, Tian Y, et al. Hospital variation in mortality and failure to rescue after surgery for high-risk neonatal diagnoses. Neonatology 2024;121:34-45.
7. Benachi A, Dommergues M, Delezoide AL, et al. Tracheal obstruction in experimental diaphragmatic hernia: An endoscopic approach in the fetal lamb. Prenat Diagn 1997;17:629-34.
8. Deprest JA, Nicolaides KH, Benachi A, et al. TOTAL trial for severe hypoplasia investigators. Randomized trial of fetal surgery for severe left diaphragmatic hernia. N Engl J Med 2021;385:107-18.
9. Deprest JA, Benachi A, Gratacos E, et al. TOTAL trial for moderate hypoplasia investigators. Randomized trial of fetal surgery for moderate left diaphragmatic hernia. N Engl J Med 2021;385:119-29.
10. Russo FM, Cordier AG, Basurto D, et al. Fetal endoscopic tracheal occlusion reverses the natural history of right-sided congenital diaphragmatic hernia: European multicenter experience. Ultrasound Obstet Gynecol 2021;57:378-85.
11. Harrison MR, Adzick NS, Bullard KM, et al. Correction of congenital diaphragmatic hernia in utero VII: A prospective trial. J Pediatr Surg 1997;32:1637-42.
12. Benachi A, Delezoide AL, Chailley-Heu B, et al. Ultrastructural evaluation of lung maturation in a sheep model of diaphragmatic hernia and tracheal occlusion. Am J Respir Cell Mol Biol 1999;20:805-12.
13. Jani JC, Nicolaides KH, Gratacós E, et al.Severe diaphragmatic hernia treated by fetal endoscopic tracheal occlusion. Ultrasound Obstet Gynecol 2009;34:304-10.
14. Van Calster B, Benachi A, Nicolaides KH, et al. The randomized Tracheal Occlusion To Accelerate Lung growth (TOTAL)-trials on fetal surgery for congenital diaphragmatic hernia: Reanalysis using pooled data. Am J Obstet Gynecol 2022;226:560.e1-560.e24.
15. Sananès N, Basurto D, Cordier AG, et al. Fetoscopic endoluminal tracheal occlusion with Smart-TO balloon: Study protocol to evaluate effectiveness and safety of non-invasive removal. PLoS One 2023;18:e0273878.