Sous le terme « hypertension pulmonaire » (HTP) est regroupé un ensemble de pathologies caractérisées par une élévation progressive des résistances vasculaires pulmonaires (RVP) pouvant aboutir à une insuffisance cardiaque droite.1,2 Son diagnostic est souvent tardif, du fait de symptômes non spécifiques. Cet article s’intéresse aux facteurs de prédisposition de l’HTP et à l’algorithme diagnostique proposé dans les dernières recommandations européennes des sociétés savantes de pneumologie et de cardiologie.1 Les propositions réalisées à l’occasion du dernier Congrès mondial de l’HTP en 2024 n’ont pas modifié le dépistage et le diagnostic de manière substantielle.
Épidémiologie variable selon le type d’hypertension pulmonaire
Les données épidémiologiques de l’HTP sont peu nombreuses et dépendent du type d’HTP. Il est cependant établi que le groupe 2 (HTP secondaires aux cardiopathies gauches) représente la première cause d’HTP à travers le monde et que les HTP associées aux maladies respiratoires (HTP groupe 3) constituent la première cause d’HTP précapillaire.1,2
L’HTP (tous groupes confondus) touche 1 % de la population mondiale et jusqu’à 10 % des personnes âgées de plus de 65 ans.3
L’HTP (tous groupes confondus) touche 1 % de la population mondiale et jusqu’à 10 % des personnes âgées de plus de 65 ans.3
Hypertension artérielle pulmonaire
L’épidémiologie de l’hypertension artérielle pulmonaire (HTAP), forme rare d’HTP, est quant à elle mieux connue. En Europe, sa prévalence est de 15 à 60 cas par million d’habitants, et son incidence entre 5 et 10 cas par million d’habitants par an.1,2 L’HTAP affecte plus souvent les femmes (de 60 à 80 % selon les registres) que les hommes.4-7 Paradoxalement, il a été montré que le sexe masculin est un facteur de risque de mortalité dans l’HTAP.8,9 Les deux dernières décennies ont été marquées par un changement épidémiologique majeur. En effet, alors que l’on considérait que l’HTAP touchait majoritairement les femmes jeunes (aux alentours de 35 ans) et sans comorbidités,10,11 les données actuelles situent l’âge moyen au-delà de 60 ans, avec davantage de comorbidités.7,12,13
Hypertension pulmonaire du groupe 2
L’hypertension pulmonaire associée aux cardiopathies gauches (groupe 2) représente 65 à 80 % des cas d’HTP, affectant au moins 50 % des patients porteurs d’une insuffisance cardiaque à fraction d’éjection préservée. Sa prévalence augmente avec la sévérité de la cardiopathie gauche sous-jacente.14
Hypertension pulmonaire du groupe 3
L’HTP du groupe 3, liée aux maladies respiratoires chroniques et/ou l’hypoxie, est la deuxième forme la plus fréquente d’HTP. Sa prévalence dépend de la sévérité de la pathologie respiratoire sous-jacente : 90 % des patients porteurs d’une bronchopneumopathie chronique obstructive (BPCO) GOLD IV ont une pression artérielle pulmonaire moyenne (PAPm) supérieure à 20 mmHg ; la prévalence est de 8 à 15 % dans la fibrose pulmonaire idiopathique (FPI) au bilan initial et augmente à plus de 60 % chez les patients à un stade très avancé.15 La majorité des HTP du groupe 3 sont des HTP non sévères. Seule une minorité de patients développe une HTP sévère, définie par des RVP supérieures à 5 UW.
Hypertension pulmonaire du groupe 4
Quant à l’hypertension pulmonaire thromboembolique chronique (HTP-TEC, groupe 4), son incidence et sa prévalence sont respectivement de 2 à 6 et de 26 à 38 cas par million d’habitants, avec un nombre croissant de patients diagnostiqués.2
Suspicion du diagnostic d’HTP
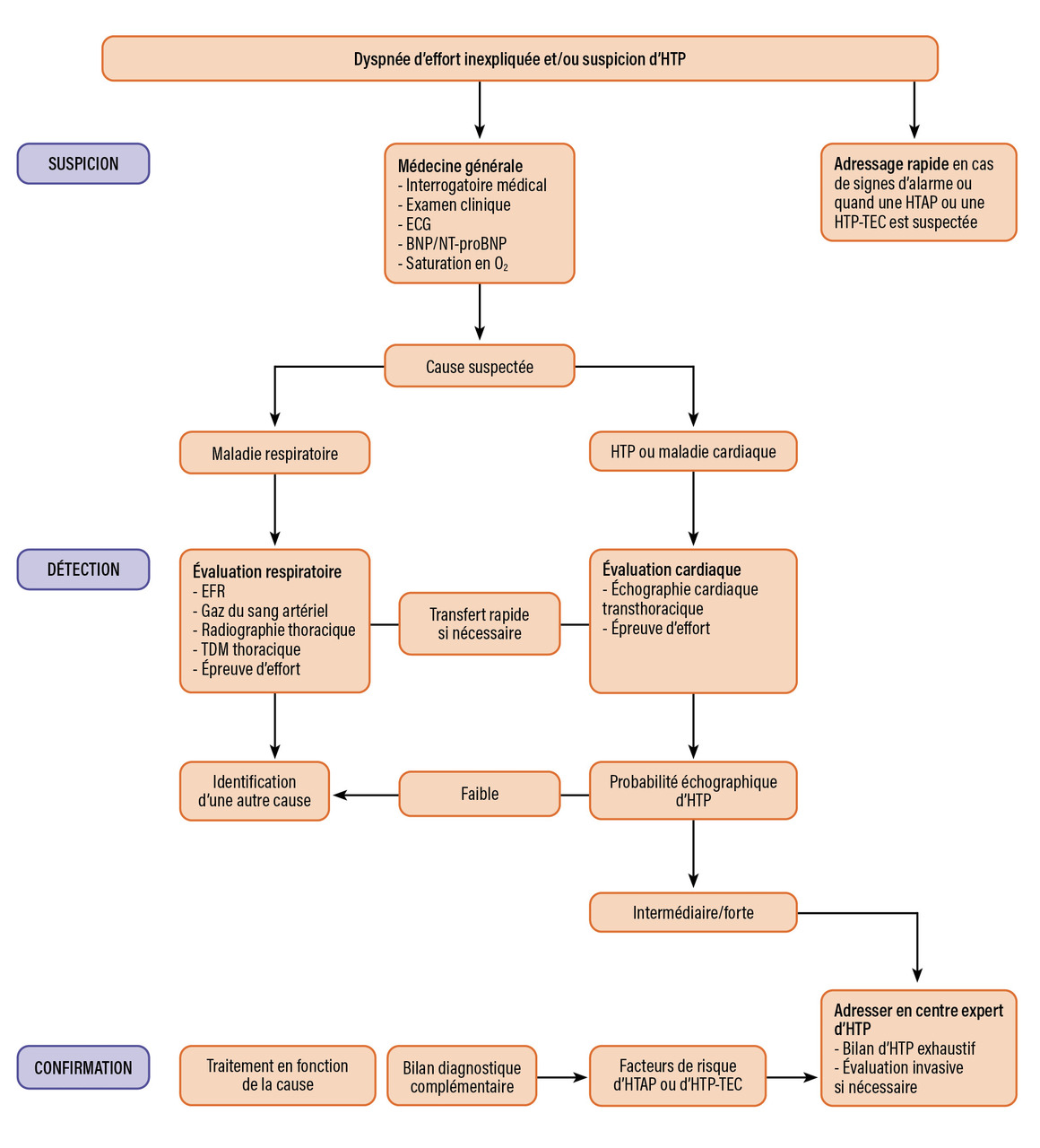
Cette première étape est le plus souvent réalisée par le médecin traitant (fig. 1 ).1,2
Interrogatoire médical
L’interrogatoire médical peut conforter la suspicion d’HTP et orienter vers la maladie associée à l’origine de cette HTP. Des facteurs prédisposant à l’HTP existent et diffèrent en fonction des groupes d’HTP.
tableau 1 ]. L’exposition aux anorexigènes, en particulier les dérivés de la fenfluramine, a été associée à de nombreux cas d’HTAP en Europe et en Amérique du Nord à la fin des années 1980 et au début des années 1990.17 Leur consommation multipliait par 6 le risque d’HTAP et par 23 en cas d’utilisation pendant plus de trois mois.17 L’incidence des HTAP liées aux anorexigènes est aujourd’hui quasiment nulle, depuis l’interdiction de ces molécules en France. Parmi les autres médicaments imputables au développement de l’HTAP, le dasatinib, un inhibiteur de tyrosine kinase (inhibiteur de Src/Abl) utilisé en deuxième ligne dans le traitement de la leucémie myéloïde chronique, est à l’origine d’HTP précapillaires sévères du point de vue clinique, fonctionnel et hémodynamique.18 L’incidence de l’HTAP est estimée à 0,45 % des patients traités par dasatinib.
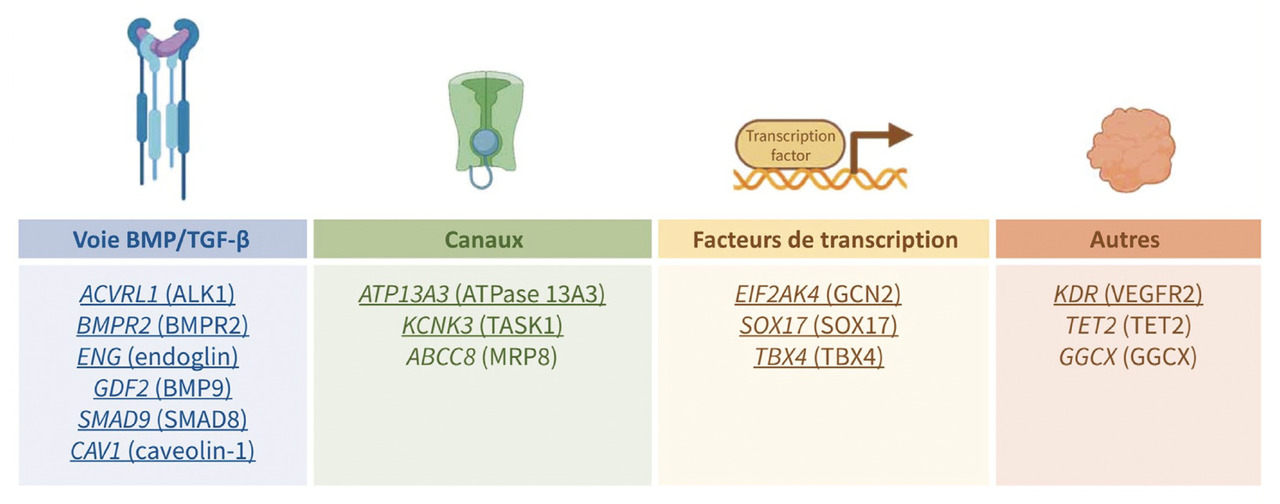
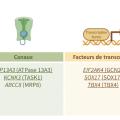
L’analyse des formes familiales d’HTAP a permis d’identifier plusieurs gènes impliqués dans l’HTAP dite héritable, dont le plus fréquent est le gène BMPR2 (bone morphogenetic protein type II). À ce jour, plus de 800 variants pathogènes différents du gène BMPR2 ont été identifiés. Les mutations du gène BMPR2 sont identifiées dans environ 75 % des formes familiales d’HTAP.16 Elles se transmettent selon un mode autosomique dominant, avec une pénétrance incomplète de l’ordre de 10 à 20 %, expliquant la possibilité de sauts de générations compliquant l’identification des formes familiales. Les gènes ACVRL1 et Endogline (ENG) sont principalement retrouvés chez les patients atteints de maladie de Rendu-Osler. Parmi les pathogènes moins fréquents, citons le variant GDF2 du gène BMP9 (bone morphogenetic protein 9), CAV1 (caveolin-1), SMAD9, KCNK3 et ATP13A3. Il existe également des formes héritables de maladies veino-occlusives secondaires à des mutations bialléliques du gène EIF2AK4 qui se transmettent selon un mode autosomique récessif1,2 et donc plus fréquemment observées dans les familles consanguines. Certains gènes sont associés à des anomalies du développement pulmonaire, comme le gène TBX4 (T-box transcription factor 4) lié au syndrome coxo-podo-patellaire (small patella syndrome), le gène KDR et d’autres associés aux cardiopathies congénitales (gène SOX17 [SRY-box transcription factor 17]). L’ensemble des principaux gènes inclus dans le panel des tests génétiques est résumé dans lafigure 2 .
Facteurs de risque médicamenteux
Plusieurs facteurs de risque médicamenteux ou toxiques ont été identifiés.1,2 Ils sont classés en deux groupes selon leur degré d’imputabilité (certain ou possible) [Facteurs de risque génétiques
Il existe également des facteurs génétiques prédisposant à l’HTAP.L’analyse des formes familiales d’HTAP a permis d’identifier plusieurs gènes impliqués dans l’HTAP dite héritable, dont le plus fréquent est le gène BMPR2 (bone morphogenetic protein type II). À ce jour, plus de 800 variants pathogènes différents du gène BMPR2 ont été identifiés. Les mutations du gène BMPR2 sont identifiées dans environ 75 % des formes familiales d’HTAP.16 Elles se transmettent selon un mode autosomique dominant, avec une pénétrance incomplète de l’ordre de 10 à 20 %, expliquant la possibilité de sauts de générations compliquant l’identification des formes familiales. Les gènes ACVRL1 et Endogline (ENG) sont principalement retrouvés chez les patients atteints de maladie de Rendu-Osler. Parmi les pathogènes moins fréquents, citons le variant GDF2 du gène BMP9 (bone morphogenetic protein 9), CAV1 (caveolin-1), SMAD9, KCNK3 et ATP13A3. Il existe également des formes héritables de maladies veino-occlusives secondaires à des mutations bialléliques du gène EIF2AK4 qui se transmettent selon un mode autosomique récessif1,2 et donc plus fréquemment observées dans les familles consanguines. Certains gènes sont associés à des anomalies du développement pulmonaire, comme le gène TBX4 (T-box transcription factor 4) lié au syndrome coxo-podo-patellaire (small patella syndrome), le gène KDR et d’autres associés aux cardiopathies congénitales (gène SOX17 [SRY-box transcription factor 17]). L’ensemble des principaux gènes inclus dans le panel des tests génétiques est résumé dans la
Recherche d’autres antécédents
Concernant l’HTP thromboembolique chronique (HTP-TEC), l’interrogatoire recherche des antécédents thromboemboliques connus (embolie pulmonaire, thrombose veineuse profonde) mais également d’autres facteurs de risque, comme la présence de matériel endovasculaire au long cours (pacemaker, Port-a-Cath), des maladies inflammatoires chroniques intestinales, une thrombocytémie essentielle, une splénectomie, un syndrome des antiphospholipides ou un antécédent de néoplasie.Symptômes et signes cliniques
Le diagnostic est souvent retardé du fait du caractère non spécifique des symptômes. La présence de signes d’insuffisance cardiaque droite et/ou les antécédents familiaux peuvent aider le médecin à évoquer ce diagnostic d’HTP.
Il n’existe aucun signe clinique spécifique de l’HTP. Les manifestations cliniques observées dans cette pathologie traduisent principalement le retentissement de la maladie sur le cœur droit ou les manifestations liées à la maladie sous-jacente (sclérodermie, cirrhose…).1,2 La dyspnée d’effort, observée dans 95 % des cas, représente la plainte la plus fréquente dans l’HTAP.1,2 L’apparition de cette dyspnée est en règle générale progressive et souvent négligée, expliquant ainsi le retard diagnostique (environ deux ans entre le début des symptômes et le diagnostic).4 Douleurs thoraciques, lipothymies et/ou syncopes peuvent survenir à l’effort. Leur présence constitue des critères de gravité de la maladie. Les palpitations sont fréquentes à l’effort et peuvent révéler des troubles du rythme cardiaque. Une dysphonie est parfois observée dans l’HTAP, témoignant d’une paralysie récurentielle gauche secondaire à la compression du nerf récurrent par le tronc de l’artère pulmonaire gauche dilatée (syndrome d’Ortner).1,2
Les signes cliniques ne sont pas spécifiques : éclat du second bruit cardiaque (B2) au foyer pulmonaire, souffle systolique d’insuffisance tricuspide. Les signes d’insuffisance cardiaque droite témoignent d’une forme avancée de la maladie (turgescence jugulaire, reflux hépatojugulaire, œdèmes des membres inférieurs et ascite).1,2
Il n’existe aucun signe clinique spécifique de l’HTP. Les manifestations cliniques observées dans cette pathologie traduisent principalement le retentissement de la maladie sur le cœur droit ou les manifestations liées à la maladie sous-jacente (sclérodermie, cirrhose…).1,2 La dyspnée d’effort, observée dans 95 % des cas, représente la plainte la plus fréquente dans l’HTAP.1,2 L’apparition de cette dyspnée est en règle générale progressive et souvent négligée, expliquant ainsi le retard diagnostique (environ deux ans entre le début des symptômes et le diagnostic).4 Douleurs thoraciques, lipothymies et/ou syncopes peuvent survenir à l’effort. Leur présence constitue des critères de gravité de la maladie. Les palpitations sont fréquentes à l’effort et peuvent révéler des troubles du rythme cardiaque. Une dysphonie est parfois observée dans l’HTAP, témoignant d’une paralysie récurentielle gauche secondaire à la compression du nerf récurrent par le tronc de l’artère pulmonaire gauche dilatée (syndrome d’Ortner).1,2
Les signes cliniques ne sont pas spécifiques : éclat du second bruit cardiaque (B2) au foyer pulmonaire, souffle systolique d’insuffisance tricuspide. Les signes d’insuffisance cardiaque droite témoignent d’une forme avancée de la maladie (turgescence jugulaire, reflux hépatojugulaire, œdèmes des membres inférieurs et ascite).1,2
Lire aussi | Consensus sur l’hypertension pulmonaire
Détection : éléments d’orientation
La deuxième étape de l’algorithme diagnostique repose sur des examens cardiorespiratoires non invasifs qui visent à écarter ou renforcer la probabilité d’HTP (fig. 1 ).1,2
Dépistage des populations à risque
Un dépistage régulier est recommandé chez les populations à risque telles que les patients atteints de sclérodermie (algorithme DETECT), porteurs du virus de l’immunodéficience humaine (VIH), les patients en attente de transplantation hépatique.1,2
Une consultation de conseil génétique et un dépistage annuel sont recommandés chez les apparentés au premier degré de patients atteints d’HTAP héritable.1,2
Enfin, il est recommandé d’explorer les patients avec dyspnée persistante ou de novo après une embolie pulmonaire.1,2
Une consultation de conseil génétique et un dépistage annuel sont recommandés chez les apparentés au premier degré de patients atteints d’HTAP héritable.1,2
Enfin, il est recommandé d’explorer les patients avec dyspnée persistante ou de novo après une embolie pulmonaire.1,2
Examens complémentaires au stade de détection
Six examens sont utiles, à ce stade.
fig. 3 ]. Elle permet aussi de rechercher des anomalies parenchymateuses témoignant d’une maladie respiratoire associée (emphysème, fibrose pulmonaire, etc.), d’une maladie veino-occlusive ou encore d’une origine post-capillaire.1,2
Outre le calcul de la probabilité échographique d’HTP, cet examen permet de détecter des signes évocateurs d’une cardiopathie gauche et orientant vers une HTP post-capillaire.
En cas de probabilité intermédiaire ou forte d’HTP, les patients doivent être orientés vers un centre expert (fig. 1 ) afin de confirmer le diagnostic et réaliser un bilan diagnostique et étiologique complémentaire.
Électrocardiogramme
L’électrocardiogramme révèle classiquement une déviation axiale droite, une hypertrophie ventriculaire droite et un bloc de branche droit.1,2Biomarqueurs
Les marqueurs cardiaques tels que le BNP (Brain natriuretic peptide) et/ou le NT-proBNP (N-terminal pro-brain natriuretic peptide) peuvent être augmentés en cas d’HTP. Ces biomarqueurs sanguins sont également utilisés dans le cadre du suivi des patients atteints d’HTP, une fois le diagnostic confirmé.Radiographie thoracique
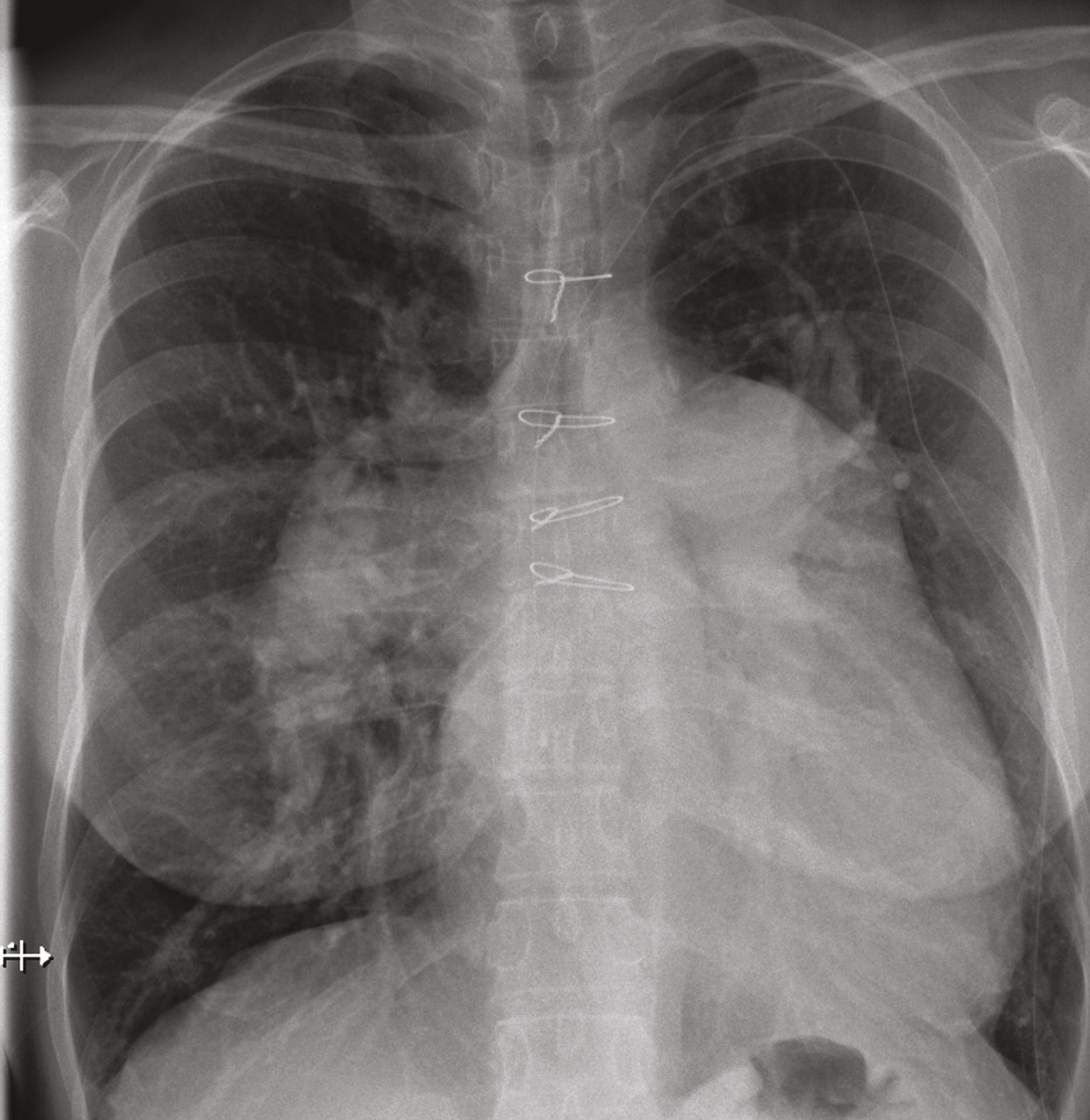
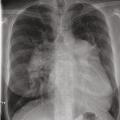
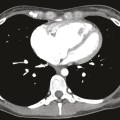
La radiographie thoracique montre généralement une dilatation des artères pulmonaires proximales et un élargissement des cavités cardiaques droites (oreillette et ventricule droit) [Épreuves fonctionnelles respiratoires
Les épreuves fonctionnelles respiratoires permettent d’identifier une pathologie respiratoire obstructive ou restrictive sous-jacente (HTP du groupe 3). Dans l’HTAP, les volumes pulmonaires et les débits sont sensiblement normaux, mais une diminution de la diffusion du monoxyde de carbone (DLCO) est quasi constante, traduisant l’atteinte vasculaire pulmonaire.19,20 Les gaz du sang en air ambiant révèlent généralement une pression artérielle en oxygène normale ou une légère hypoxémie, associée à une diminution de la pression artérielle en dioxyde de carbone (hypocapnie).1,2Épreuve d’effort
L’épreuve fonctionnelle à l’exercice est un examen utile pour explorer la dyspnée d’effort et l’intolérance à l’exercice. Il est conseillé de réaliser cet examen dans un centre expert en raison du risque potentiel d’une épreuve d’effort maximale chez les patients atteints d’HTP. Ces derniers ont généralement une pression partielle basse de dioxyde de carbone en fin d’expiration, un équivalent ventilatoire élevé pour le dioxyde de carbone, un faible pouls d’oxygène et une consommation maximale en oxygène abaissée.1,2,21Échocardiographie transthoracique
L’échocardiographie transthoracique constitue l’examen de dépistage de référence.1,2 Elle permet de déterminer une probabilité échographique d’HTP classée en trois niveaux (faible, intermédiaire et forte), fondée sur la vitesse maximale du flux de régurgitation tricuspide (VmaxIT) et d’autres signes indirects tels que la taille des cavités cardiaques droites, la collapsibilité de la veine cave inférieure, la fonction ventriculaire droite ou encore la présence d’un épanchement péricardique.1,2 Une VmaxIT inférieure ou égale à 2,8 m/s est évocatrice d’une probabilité faible d’HTP, alors qu’entre 2,8 et 3,4 m/s on considère que la probabilité est intermédiaire. La présence d’autres signes échographiques évocateurs d’HTP renforce la probabilité échographique du patient (de faible à intermédiaire ou d’intermédiaire à élevée). Une VmaxIT supérieure à 3,4 m/s est, quant à elle, évocatrice d’une probabilité élevée d’HTP, quels que soient les signes échographiques associés.Outre le calcul de la probabilité échographique d’HTP, cet examen permet de détecter des signes évocateurs d’une cardiopathie gauche et orientant vers une HTP post-capillaire.
En cas de probabilité intermédiaire ou forte d’HTP, les patients doivent être orientés vers un centre expert (
Lire aussi | Hypertension pulmonaire : les 10 messages-clés
Confirmation diagnostique
Cathétérisme cardiaque droit : l’examen de référence
Le cathétérisme cardiaque droit est le seul examen permettant de confirmer le diagnostic d’HTP. Cet examen doit être réalisé dans un centre expert.1,2
Il est recommandé de référer rapidement les patients présentant des signes d’alerte vers un centre expert. Ces signes d’alerte sont les suivants : symptômes sévères ou évoluant rapidement (classe fonctionnelle de la New York Heart Association [NYHA] III/IV), signes cliniques d’insuffisance cardiaque droite, syncope, signes de bas débit cardiaque, arythmies mal tolérées et état hémodynamique altéré (hypotension, tachycardie) [fig. 1 ].
L’atteinte précapillaire de l’HTP est définie par une pression artérielle pulmonaire (PAP) moyenne supérieure à 20 mmHg au repos, une pression artérielle pulmonaire d’occlusion (reflet de la pression capillaire) inférieure ou égale à 15 mmHg et des RVP supérieures à 2 UW.1,2 La réalisation d’un test de remplissage par administration rapide d’une solution saline peut démasquer une dysfonction cardiaque gauche.1,2
Dans le cas d’une HTAP idiopathique, héritable ou secondaire à la prise d’anorexigènes, il est nécessaire d’effectuer un test de vasodilatation par le monoxyde d’azote (NO) inhalé.1,2 Ce test permet, lorsqu’il est positif (diminution de la PAP moyenne de plus de 10 mmHg avec une PAP moyenne inférieure ou égale à 40 mmHg et un débit cardiaque normal ou élevé), d’identifier un sous-groupe de patients ayant un meilleur pronostic et pouvant bénéficier d’un traitement prolongé par inhibiteurs calciques.1,2,22
Il est recommandé de référer rapidement les patients présentant des signes d’alerte vers un centre expert. Ces signes d’alerte sont les suivants : symptômes sévères ou évoluant rapidement (classe fonctionnelle de la New York Heart Association [NYHA] III/IV), signes cliniques d’insuffisance cardiaque droite, syncope, signes de bas débit cardiaque, arythmies mal tolérées et état hémodynamique altéré (hypotension, tachycardie) [
L’atteinte précapillaire de l’HTP est définie par une pression artérielle pulmonaire (PAP) moyenne supérieure à 20 mmHg au repos, une pression artérielle pulmonaire d’occlusion (reflet de la pression capillaire) inférieure ou égale à 15 mmHg et des RVP supérieures à 2 UW.1,2 La réalisation d’un test de remplissage par administration rapide d’une solution saline peut démasquer une dysfonction cardiaque gauche.1,2
Dans le cas d’une HTAP idiopathique, héritable ou secondaire à la prise d’anorexigènes, il est nécessaire d’effectuer un test de vasodilatation par le monoxyde d’azote (NO) inhalé.1,2 Ce test permet, lorsqu’il est positif (diminution de la PAP moyenne de plus de 10 mmHg avec une PAP moyenne inférieure ou égale à 40 mmHg et un débit cardiaque normal ou élevé), d’identifier un sous-groupe de patients ayant un meilleur pronostic et pouvant bénéficier d’un traitement prolongé par inhibiteurs calciques.1,2,22
Bilan à visée étiologique
Biologique
Dans le cadre du bilan étiologique de l’HTAP, un bilan biologique est nécessaire, comprenant les sérologies pour le VIH et les hépatites virales B et C ainsi que les marqueurs d’auto-immunité, notamment à la recherche d’une sclérodermie ou d’un lupus.1,2Imagerie
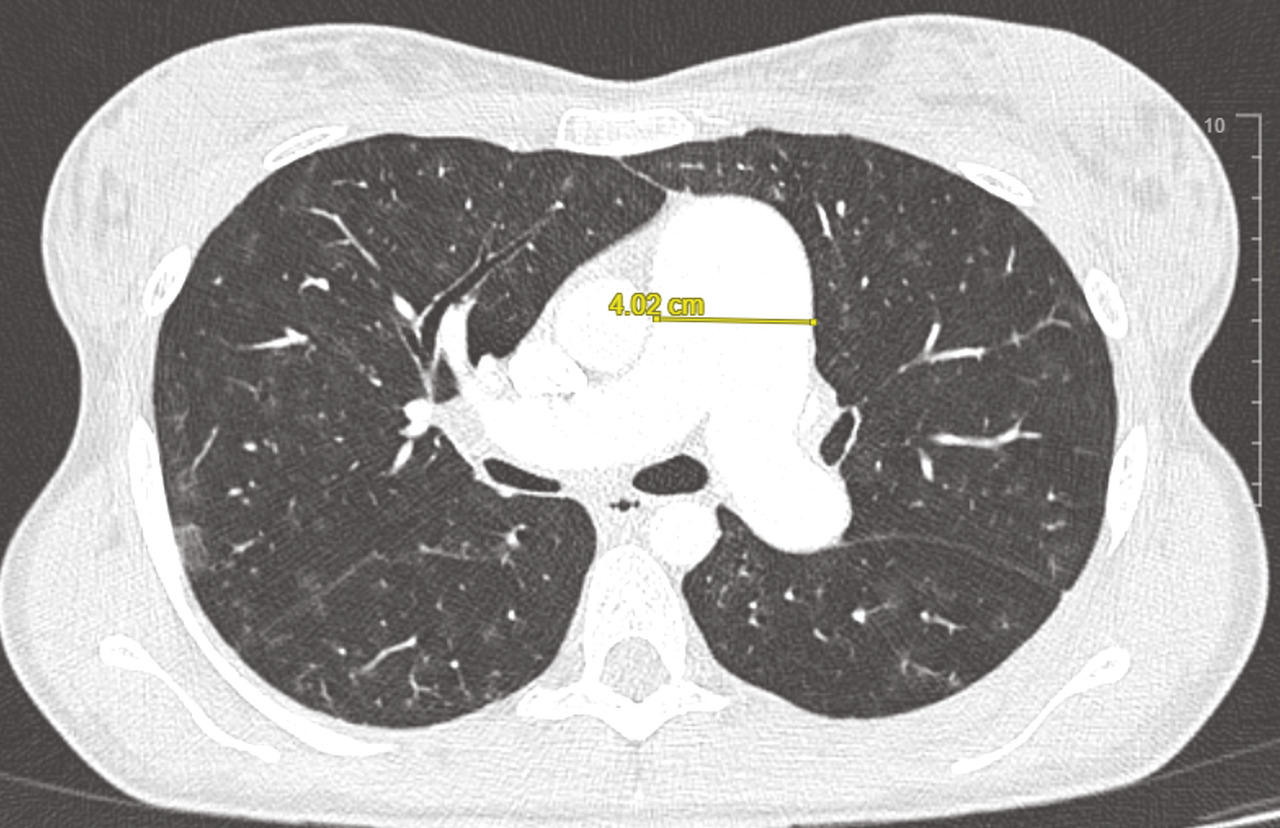
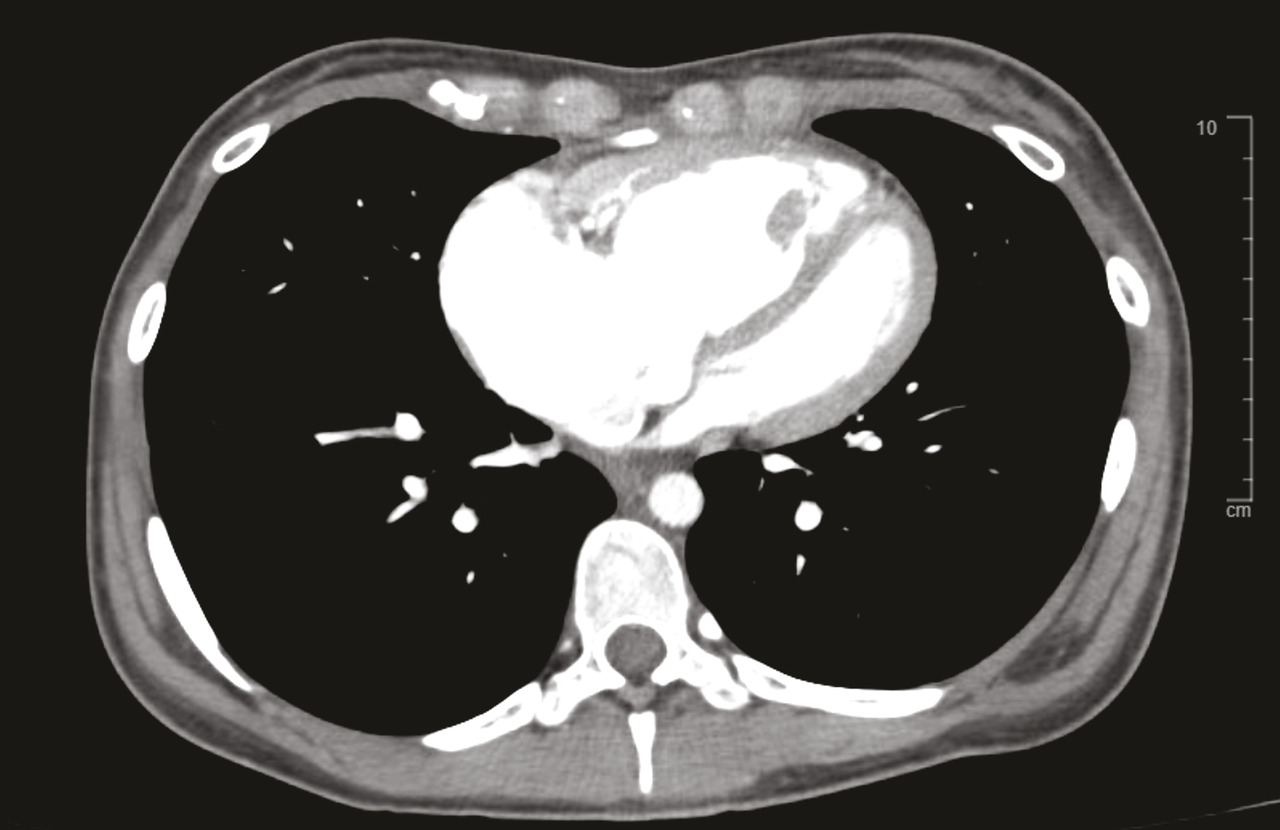
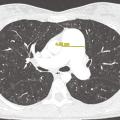
Le scanner thoracique en coupes millimétriques permet de rechercher des éléments évocateurs de maladie veino-occlusive pulmonaire (épaississement des septa, nodules flous en verre dépoli, adénopathies médiastinales) ou d’une pathologie respiratoire sous-jacente (emphysème, pneumopathie interstitielle diffuse…) [fig. 4 et 5].1,2Un angioscanner thoracique est indispensable pour le bilan d’opérabilité des patients atteints d’HTP-TEC.
L’injection de produit de contraste au temps artériel pulmonaire (angio-TDM thoracique) permet de rechercher des lésions thromboemboliques chroniques.
En cas de doute, la scintigraphie pulmonaire de ventilation et de perfusion est l’examen de référence pour confirmer, ou infirmer, ce diagnostic (défects perfusionnels non concordants avec la ventilation).1,2
Bien que de moins en moins utilisée, l’angiographie pulmonaire peut être réalisée dans le cadre du bilan thérapeutique (avant thrombo-endartériectomie et/ou angioplasties pulmonaires) chez les patients atteints d’HTP-TEC.1,2
L’échographie abdominale avec Doppler du tronc porte est utile pour rechercher une hypertension portale. Les signes échographiques recherchés sont l’augmentation du calibre des veines porte et splénique, la présence d’ascite, une splénomégalie, les voies de dérivation porto-cave.
HTP confirmée : évaluer sa sévérité
Les différentes variables utilisées pour évaluer la sévérité de l’HTP ont été validées, essentiellement dans l’HTAP. Leur caractérisation permet de mettre en place la stratégie thérapeutique la plus adaptée.
Variables cliniques
La dyspnée s’apprécie à l’aide de l’interrogatoire du patient et de la classe fonctionnelle NYHA (tableau 2 ). Malgré son évaluation subjective et sa variabilité interopérateurs, cette classification demeure l’un des outils pronostiques les plus puissants dans l’HTP.1,2
Capacité d’exercice
Le test de marche de six minutes, facile à réaliser, rapide et non invasif, constitue une pierre angulaire de l’évaluation pronostique des HT(A)P.1,2 La distance parcourue au cours de ce test a longtemps été utilisée comme critère de jugement principal des essais thérapeutiques menés dans l’HTAP.
Biomarqueurs pronostiques
Aucun biomarqueur n’est spécifique de l’HTAP ou du remodelage vasculaire. Bien que de nombreux biomarqueurs pronostiques aient été identifiés, peu sont utilisés en routine. Il s’agit principalement de marqueurs de la fonction ventriculaire droite (le BNP et le NT-proBNP) et de son retentissement sur les autres organes (bilirubine, urée, créatinine).1,2
Hémodynamique
Certaines variables hémodynamiques mesurées lors du cathétérisme cardiaque droit apportent des informations pronostiques, telles que la pression auriculaire droite, l’index cardiaque, le volume d’éjection systolique indexé et la saturation en oxygène du sang veineux mêlé (SvO2).1,2
L’imagerie par résonance magnétique (IRM) cardiaque est de plus en plus utilisée à travers le monde pour évaluer la taille, la morphologie et la fonction ventriculaire droite de façon non invasive.1,2 En revanche, cet examen ne permet pas la mesure précise de la pression artérielle pulmonaire. Il ne peut donc pas, à l’heure actuelle, se substituer au cathétérisme cardiaque droit.
L’imagerie par résonance magnétique (IRM) cardiaque est de plus en plus utilisée à travers le monde pour évaluer la taille, la morphologie et la fonction ventriculaire droite de façon non invasive.1,2 En revanche, cet examen ne permet pas la mesure précise de la pression artérielle pulmonaire. Il ne peut donc pas, à l’heure actuelle, se substituer au cathétérisme cardiaque droit.
Réduire l’errance diagnostique
Le diagnostic précoce de l’HTP demeure un défi et repose sur de nombreux professionnels de santé, du médecin généraliste pour la suspicion au diagnostic définitif en centre expert. Du fait des nombreuses causes de l’HTP, une collaboration étroite entre la médecine de ville et de spécialité est nécessaire afin de réduire au minimum l’errance diagnostique de ces patients.
Références
1. Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 2023;61(1):2200879.
2. Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2022;43(38):3618‑731.
3. Hoeper MM, Humbert M, Souza R, et al. A global view of pulmonary hypertension. Lancet Respir Med 2016;4(4):306‑22.
4. Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France. Am J Respir Crit Care Med 2006;173(9):1023‑30.
5. Badesch DB, Raskob GE, Elliott CG, et al. Pulmonary arterial hypertension: Baseline characteristics from the REVEAL Registry. Chest 2010;137(2):376‑87.
6. Peacock AJ, Murphy NF, McMurray JJV. An epidemiological study of pulmonary arterial hypertension. Eur Respir J 2007;30(1):104‑9.
7. Ling Y, Johnson MK, Kiely DG, et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: Results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med 2012;186(8):790‑6.
8. Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010;122(2):156‑63.
9. Benza RL, Gomberg-Maitland M, Miller DP, et al. The REVEAL Registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest 2012;141(2):354‑62.
10. D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension results from a national prospective registry. Ann Intern Med 1991;115(5):343‑9.
11. Rich S, Dantzker DR, Ayres SM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med 1987;107(2):216‑23.
12. Boucly A, Savale L, Jais X, et al. Association between initial treatment strategy and long-term survival in pulmonary arterial hypertension. Am J Respir Crit Care Med 2021;204(7):842‑54.
13. Hoeper MM, Kramer T, Pan Z, et al. Mortality in pulmonary arterial hypertension: Prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur Respir J 2017;50(2):1700740.
14. Rosenkranz S, Gibbs JSR, Wachter R, et al. Left ventricular heart failure and pulmonary hypertension. Eur Heart J 2016;37(12):942‑54.
15. Nathan SD, Barbera JA, Gaine SP, et al. Pulmonary hypertension in chronic lung disease and hypoxia. Eur Respir J 2019;53(1):1801914.
16. Soubrier F, Chung WK, Machado R, et al. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol 2013;62(25 Suppl):D13-21.
17. Abenhaim L, Moride Y, Brenot F, et al. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N Engl J Med 1996;335(9):609‑16.
18. Montani D, Bergot E, Günther S, et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation 2012;125(17):2128‑37.
19. Sun XG, Hansen JE, Oudiz RJ, et al. Pulmonary function in primary pulmonary hypertension. J Am Coll Cardiol 2003;41(6):1028‑35.
20. Trip P, Nossent EJ, de Man FS, et al. Severely reduced diffusion capacity in idiopathic pulmonary arterial hypertension: Patient characteristics and treatment responses. Eur Respir J 2013;42(6):1575‑85.
21. Sun XG, Hansen JE, Oudiz RJ, et al. Exercise pathophysiology in patients with primary pulmonary hypertension. Circulation 2001;104(4):429‑35.
22. Sitbon O, Humbert M, Jagot JL, et al. Inhaled nitric oxide as a screening agent for safely identifying responders to oral calcium-channel blockers in primary pulmonary hypertension. Eur Respir J 1998;12(2):265‑70.
23. Eichstaedt CA, Belge C, Chung WK, et al. Genetic counselling and testing in pulmonary arterial hypertension: A consensus statement on behalf of the International Consortium for Genetic Studies in PAH. Eur Respir J 2023;61(2):2201471.
2. Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2022;43(38):3618‑731.
3. Hoeper MM, Humbert M, Souza R, et al. A global view of pulmonary hypertension. Lancet Respir Med 2016;4(4):306‑22.
4. Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France. Am J Respir Crit Care Med 2006;173(9):1023‑30.
5. Badesch DB, Raskob GE, Elliott CG, et al. Pulmonary arterial hypertension: Baseline characteristics from the REVEAL Registry. Chest 2010;137(2):376‑87.
6. Peacock AJ, Murphy NF, McMurray JJV. An epidemiological study of pulmonary arterial hypertension. Eur Respir J 2007;30(1):104‑9.
7. Ling Y, Johnson MK, Kiely DG, et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: Results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med 2012;186(8):790‑6.
8. Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010;122(2):156‑63.
9. Benza RL, Gomberg-Maitland M, Miller DP, et al. The REVEAL Registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest 2012;141(2):354‑62.
10. D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension results from a national prospective registry. Ann Intern Med 1991;115(5):343‑9.
11. Rich S, Dantzker DR, Ayres SM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med 1987;107(2):216‑23.
12. Boucly A, Savale L, Jais X, et al. Association between initial treatment strategy and long-term survival in pulmonary arterial hypertension. Am J Respir Crit Care Med 2021;204(7):842‑54.
13. Hoeper MM, Kramer T, Pan Z, et al. Mortality in pulmonary arterial hypertension: Prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur Respir J 2017;50(2):1700740.
14. Rosenkranz S, Gibbs JSR, Wachter R, et al. Left ventricular heart failure and pulmonary hypertension. Eur Heart J 2016;37(12):942‑54.
15. Nathan SD, Barbera JA, Gaine SP, et al. Pulmonary hypertension in chronic lung disease and hypoxia. Eur Respir J 2019;53(1):1801914.
16. Soubrier F, Chung WK, Machado R, et al. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol 2013;62(25 Suppl):D13-21.
17. Abenhaim L, Moride Y, Brenot F, et al. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N Engl J Med 1996;335(9):609‑16.
18. Montani D, Bergot E, Günther S, et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation 2012;125(17):2128‑37.
19. Sun XG, Hansen JE, Oudiz RJ, et al. Pulmonary function in primary pulmonary hypertension. J Am Coll Cardiol 2003;41(6):1028‑35.
20. Trip P, Nossent EJ, de Man FS, et al. Severely reduced diffusion capacity in idiopathic pulmonary arterial hypertension: Patient characteristics and treatment responses. Eur Respir J 2013;42(6):1575‑85.
21. Sun XG, Hansen JE, Oudiz RJ, et al. Exercise pathophysiology in patients with primary pulmonary hypertension. Circulation 2001;104(4):429‑35.
22. Sitbon O, Humbert M, Jagot JL, et al. Inhaled nitric oxide as a screening agent for safely identifying responders to oral calcium-channel blockers in primary pulmonary hypertension. Eur Respir J 1998;12(2):265‑70.
23. Eichstaedt CA, Belge C, Chung WK, et al. Genetic counselling and testing in pulmonary arterial hypertension: A consensus statement on behalf of the International Consortium for Genetic Studies in PAH. Eur Respir J 2023;61(2):2201471.
Dans cet article

Résumé
Le diagnostic de l’hypertension pulmonaire est souvent tardif du fait de la faible spécificité des signes cliniques dominés par la dyspnée d’effort. Les recommandations européennes des sociétés savantes de pneumologie et de cardiologie, publiées en 2022, ont permis de préciser l’algorithme diagnostique. Le diagnostic de l’hypertension pulmonaire repose sur une approche en trois étapes. La première, « suspicion », est fondée principalement sur les symptômes et les signes cliniques présentés par le patient. La deuxième, « détection », repose sur des examens paracliniques de dépistage tels que l’échographie cardiaque qui permettent d’écarter ou de renforcer la suspicion diagnostique. Enfin, si la probabilité échographique d’hypertension pulmonaire est élevée, la troisième étape vise à confirmer le diagnostic par la réalisation d’un cathétérisme cardiaque droit.