La mucoviscidose est la plus fréquente des maladies génétiques autosomiques récessives létales dans la population caucasienne. En France, sa fréquence est de 1/4 500 naissances, et la proportion des sujets hétérozygotes porteurs sains est estimée à 1/25. La survie des patients s’est considérablement améliorée, avec une accélération récente grâce à l’introduction des modulateurs de CFTR.1 La médiane de survie d’un enfant né en 2024 est désormais considérée comme équivalente à celle de la population générale.2
Plus de 2 000 mutations génétiques identifiées
La mucoviscidose est due à des mutations du gène CFTR (cystic fibrosis transmembrane conductance regulator) situé sur le bras long du chromosome 7 et codant pour une protéine transmembranaire intervenant dans la régulation du transport transépithélial des ions chlorure (Cl-). L’absence ou la dysfonction de la protéine CFTR entraîne un défaut du transport du Cl-, donc une augmentation de la réabsorption de sel et d’eau et, notamment au niveau de l’épithélium bronchique, une réduction du liquide de surface bronchique.
Il s’agit d’une exocrinopathie généralisée touchant les glandes séreuses et les glandes à sécrétion muqueuse qui entraîne une accumulation de sécrétions visqueuses et déshydratées.
Ce « mucus visqueux » (d’où le nom de mucoviscidose) obstrue différents sites de l’organisme, notamment l’appareil respiratoire, le tube digestif et ses annexes (pancréas, voies biliaires et foie), les glandes sudoripares et le tractus génital.
Plus de 2 000 mutations ont été identifiées à ce jour. Elles peuvent être soit identiques sur les deux allèles (homozygotie), soit différentes (hétérozygotie composite). En France, la mutation la plus fréquente, la délétion du 508e acide aminé (phénylalanine), est appelée F508del.
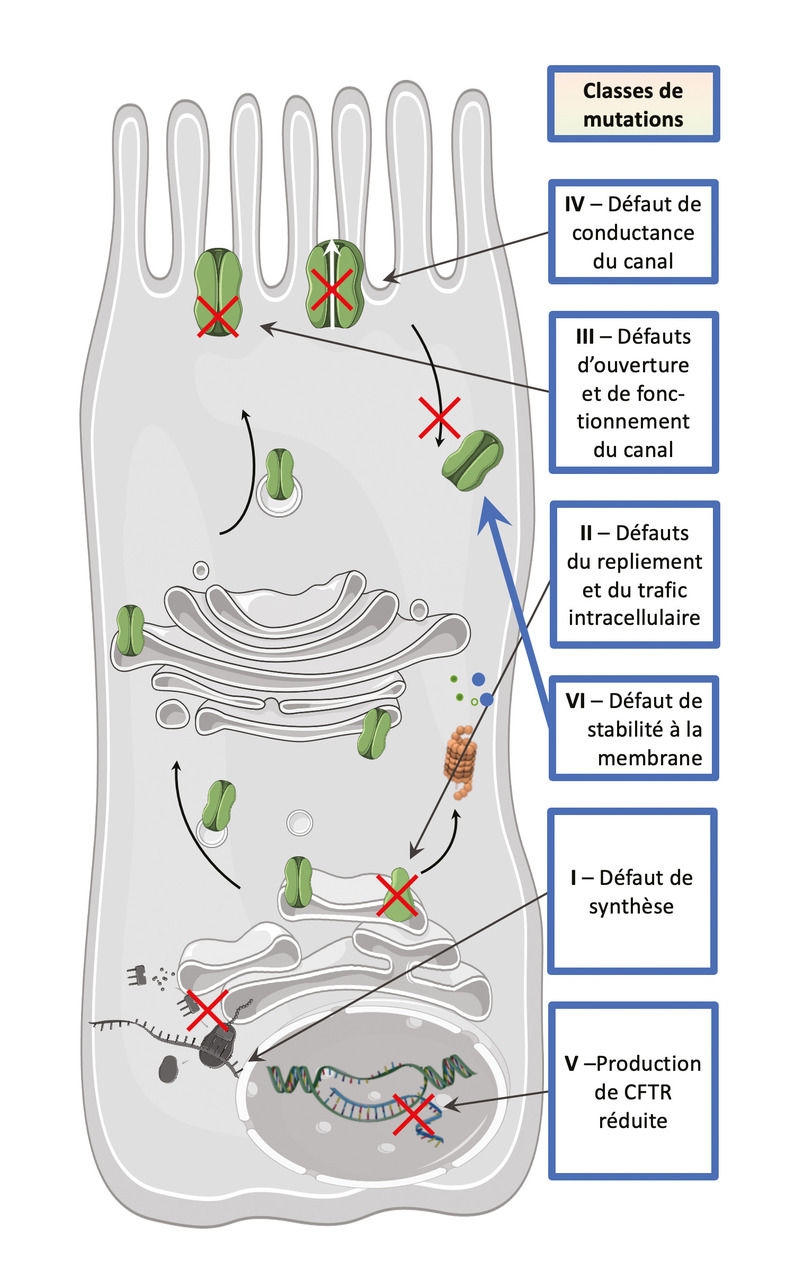
Les mutations sont classées en six catégories selon leurs conséquences sur la production, le routage, le fonctionnement ou la stabilité du canal CFTR à la membrane épithéliale (fig. 1). Les mutations de classe 1 sont des mutations de production qui ne permettent pas la synthèse de la protéine. La mutation la plus commune portée par environ 85 % des patients, F508del, appartient à la classe 2 : la protéine est synthétisée mais défectueuse, du fait d’un défaut de repliement, et donc éliminée par le protéasome. Les protéines issues de mutations de classe 3, dites de portail, atteignent la membrane mais présentent une fréquence d’ouverture du canal réduite due à un défaut d’activation. Les mutations des classes 1 à 3 conduisent à une activité de CFTR minime, voire nulle, et sont associées à des manifestations cliniques sévères. Les protéines issues des mutations de classe 4 à 6 entraînent une diminution du transport ionique (classe 4), de la synthèse protéique (classe 5) ou une demi-vie membranaire réduite (classe 6). Dans ces cas, le CFTR présente une fonction résiduelle qui conduit à des phénotypes moins graves. En général, les mutations de classe 1 ne sont pas accessibles à la thérapie protéique alors qu’un certain nombre de mutations des autres classes, dont la plus fréquente, sont accessibles à ces traitements modulateurs de CFTR.
Dépistage néonatal depuis 2002
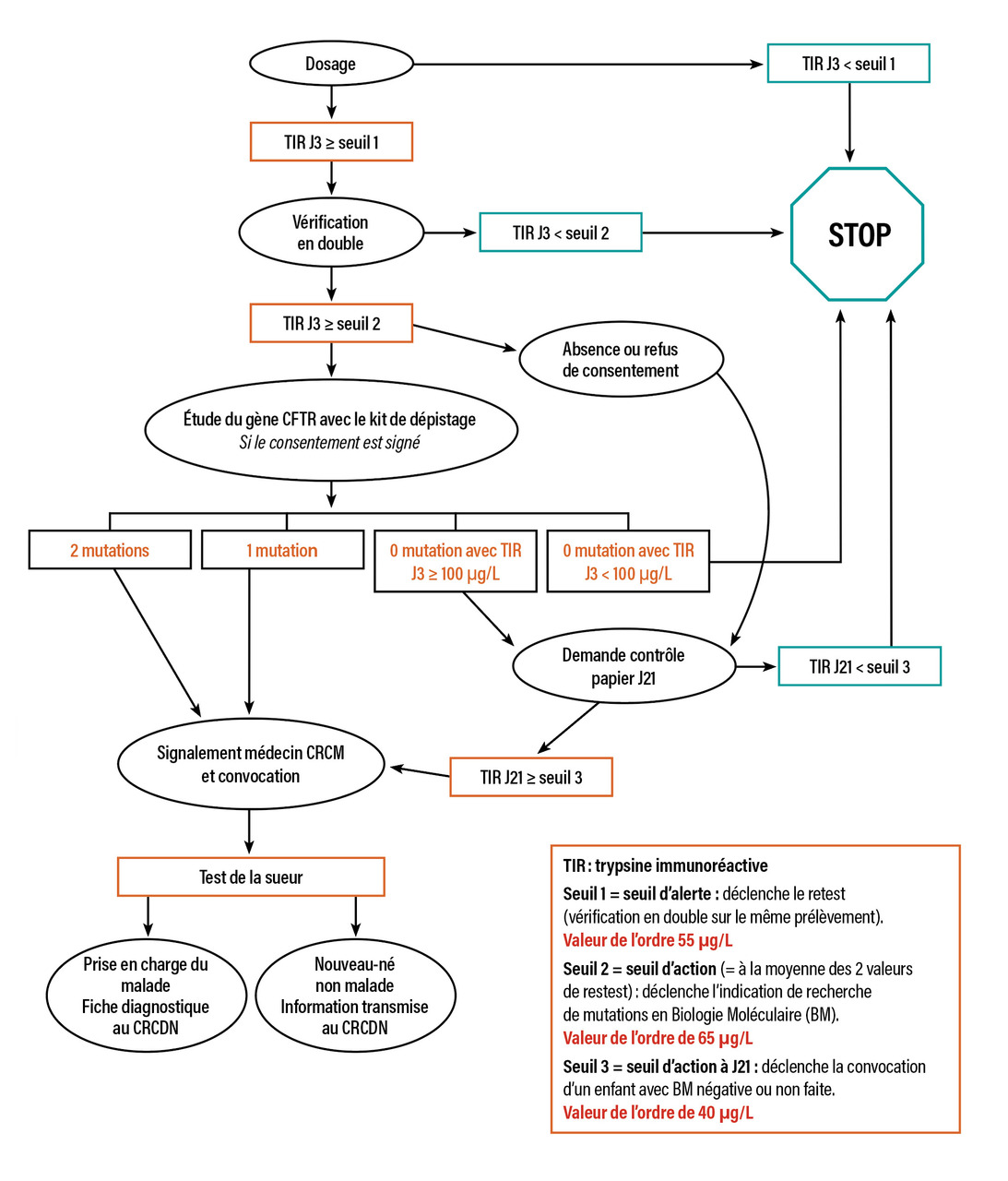
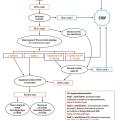
Le dépistage néonatal de la mucoviscidose est généralisé en France depuis 2002.3 Il est réalisé à partir du sang recueilli sur papier buvard pour le test de Guthrie, idéalement à 72 heures de vie, après recueil du consentement signé des parents (pour la recherche génétique éventuelle). Il repose sur le dosage de la trypsine immuno-réactive (TIR), enzyme pancréatique dont un taux élevé reflète une souffrance pancréatique. Un dosage élevé de TIR à 72 heures de vie conduit, après contrôle, à la pratique d’une recherche génétique.4 Celle-ci est fondée sur la détection d’allèles mutés sur une trousse des 29 mutations les plus fréquentes. En cas de mutation identifiée sur les deux allèles (enfant malade) ou d’un des deux allèles (situation soit de simple hétérozygotie, soit de maladie avec une deuxième mutation non identifiée), l’enfant est convoqué au centre de ressources et de compétences de la mucoviscidose (CRCM) régional afin de réaliser un test de la sueur. Si celui-ci est positif, le diagnostic de mucoviscidose est affirmé. En l’absence de mutation retrouvée, l’enfant est prélevé de nouveau à l’âge de 3 semaines pour un nouveau dosage de la TIR. Si ce dosage de TIR reste élevé, un test de la sueur est réalisé (fig. 2).
Suivi du nourrisson dépisté : mettre en place prévention et traitements
Le dépistage néonatal offre l’opportunité d’améliorer le pronostic de la maladie grâce à un suivi médical précoce, la mise en route de mesures préventives et de traitements appropriés pour retarder l’atteinte pulmonaire ou nutritionnelle. Les recommandations sont actuellement fondées sur des consensus d’experts.5
Le bilan initial est réalisé immédiatement après le diagnostic. Il comporte un interrogatoire précis sur l’état respiratoire, une évaluation nutritionnelle, un dosage de l’élastase fécale et, de façon conjointe à l’évaluation kinésithérapeutique de l’encombrement, une bactériologie des sécrétions, qui peut être réalisée par crachat induit.
Les parents rencontrent l’équipe du CRCM, le kinésithérapeute, l’infirmier coordinateur, le diététicien, le psychologue et l’assistant social. La prise en charge multidisciplinaire coordonnée par le CRCM, en lien avec le médecin traitant, est mise en place : administration d’une opothérapie pancréatique (extraits pancréatiques), de compléments sodés et kinésithérapie respiratoire. Un programme d’éducation thérapeutique est débuté.
Les études évaluant ces programmes après une à deux décennies confirment un impact favorable sur la nutrition et l’évolution respiratoire.6 - 9 Cependant, malgré cette prise en charge précoce, la maladie évolue et s’installe.10 C’est pourquoi il est envisagé de commencer les traitements modulateurs de CFTR le plus tôt possible (en fonction de l’évolution des autorisations de mise sur le marché et des accès précoces).
Ce mode de dépistage est assez sensible mais peu spécifique. On estime à environ 10 % le nombre de faux positifs de ce dépistage. En effet, la TIR peut s’élever de façon non spécifique à l’occasion de tout stress néonatal. Ces résultats faussement positifs avec un diagnostic secondairement infirmé (TIR élevée de façon non spécifique et test de la sueur négatif) sont source d’anxiété parentale.11
Diagnostic non conclu, une prise en charge particulière
Des cas non conclus sont également générés par cette stratégie (test de la sueur intermédiaire, génétique non contributive).12
Ces situations regroupent les nourrissons avec une hypertrypsinémie et présentant :
- une concentration sudorale de Cl- entre 30 et 59 mmol/L et au plus un variant du gène CFTR associé à la mucoviscidose ;
- ou une concentration sudorale de Cl- inférieure à 30 mmol/L et deux variants du gène CFTR, dont au moins un est de pathogénicité indéterminée.
Leur prévalence est variable selon les populations et l’algorithme utilisé, allant de 1 à 6 % en Australie et au Canada, jusqu’à 21 % en Californie où l’analyse génétique couvre tous les gènes. En France, depuis le retrait de la mutation R117H du kit de dépistage néonatal, ce ratio est de 11 %.
Il est important de comprendre que ces situations ne correspondent pas à un diagnostic. Elles sont révélées par une anomalie biologique transitoire, l’hypertrypsinémie, chez des nourrissons asymptomatiques. Leur prise en charge a fait l’objet de recommandations nationales spécifiques pour le diagnostic et le suivi des nourrissons.13
La plupart de ces bébés ne présenteront jamais de signe de mucoviscidose, mais certains – a priori une très faible proportion – développeront une atteinte clinique souvent révélée à l’adolescence ou à l’âge adulte (affection liée à CFTR, AL-CFTR). À l’échelon individuel, ces patients ne développeront ni mucoviscidose ni AL-CFTR dans l’immense majorité des cas. Une faible fraction de patients, porteurs de mutants rares associés à la mucoviscidose, non repérés par les kits utilisés lors du dépistage, sont à reclasser comme atteints de mucoviscidose. D’autres, une minorité, porteurs d’un génotype compatible avec une AL-CFTR, peuvent développer des symptômes le plus souvent modérés, à l’âge adulte.14 - 16 Ainsi sur 2 441 enfants dépistés positifs en France entre 2002 et 2017, 273 ont un génotype compatible avec une AL-CFTR. Une étude est en cours pour déterminer leur évolution.
Repérer les formes susceptibles d’évoluer vers une AL-CFTR, même tardivement, est un enjeu important. L’impossibilité de statuer sur l’avenir de leur enfant génère une angoisse importante chez les parents et soulève, en cas de désir de nouvelle grossesse, la question de l’éventualité du diagnostic prénatal. Ce dilemme est une question éthique majeure entre « manquer le diagnostic » d’un enfant qui sera diagnostiqué tardivement avec une maladie déjà évoluée et « classer malade » un sujet qui ne développera aucun signe de la maladie.
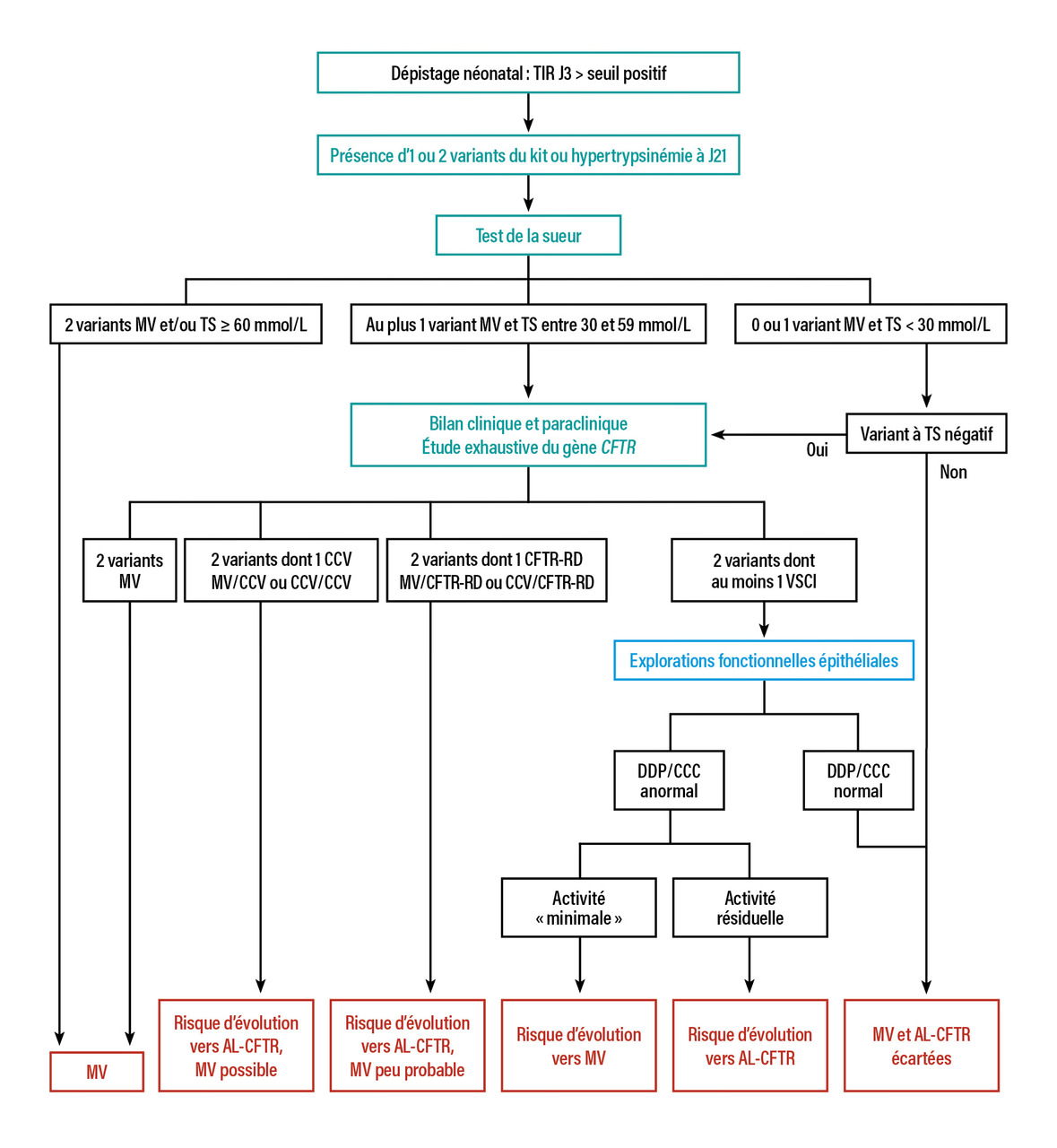
À l’échelon individuel, il n’est pas évident de pouvoir identifier ceux qui développeront une mucoviscidose. Les nourrissons avec un test de la sueur initial intermédiaire semblent plus à risque de développer une mucoviscidose par rapport à ceux qui ont un test de la sueur normal. Aucune étude n’a à ce jour caractérisé le risque de ces enfants d’évoluer vers une AL-CFTR bien que certains nourrissons présentent des génotypes compatibles. Cette classification est d’autant plus compliquée que, dans cette population, on ne peut pas parler d’AL-CFTR proprement dite mais plutôt de risque évolutif car ces nourrissons sont asymptomatiques. Ainsi l’algorithme décisionnel (fig. 3) vise à préciser le risque évolutif ultérieur (probablement à l’âge adulte) vers une AL-CFTR et, parmi ceux-ci, ceux qui sont à plus haut risque de développer une atteinte pluri-organes symptomatique.17,18 Il convient dans ce cas d’adresser à des centres spécialisés.
Le diagnostic de mucoviscidose sur symptômes est devenu rare
La généralisation du dépistage néonatal en 2002 permet que tous les enfants nés sur le territoire français bénéficient de ce dépistage. Cela implique que le diagnostic de mucoviscidose sur symptômes ne concerne que les enfants ayant présenté un résultat faussement négatif ou les enfants nés hors du territoire national. Ces formes principalement diagnostiquées chez le grand enfant ou l’adolescent ont une présentation similaire à celles de l’adulte : bronchopathie chronique évoluant vers des dilatations des bronches, pancréatite chronique ou récidivante, atteinte sinusienne chronique ; chez l’homme, atrésie des canaux déférents et, de façon plus rare, épisodes inexpliqués de déshydratation.19
Changement de paradigme grâce au dépistage néonatal
Le dépistage néonatal de la mucoviscidose a permis d’améliorer le pronostic des patients atteints de mucoviscidose. À l’ère des thérapies modulatrices de CFTR, qui révolutionnent la prise en charge des patients, un changement de paradigme peut être envisagé : que la mucoviscidose ne soit plus une maladie mortelle, grâce au diagnostic néonatal et à un traitement modulateur de CFTR permettant d’empêcher l’évolution de l’atteinte en particulier pulmonaire.
2. Lopez A, Daly C, Vega-Hernandez G, et al. Elexacaftor/tezacaftor/ivacaftor projected survival and long-term health outcomes in people with cystic fibrosis homozygous for F508del. J Cyst Fibros 2023;22:607-14.
3. Travert G, Heeley M, Heeley A. History of newborn screening for cystic fibrosis-the early years. Int J Neonatal Screen 2020;6(1):8.
4. Munck A, Cheillan D, Audrezet MP, et al. Dépistage néonatal de la mucoviscidose en France. Med Sci (Paris) 2021;37(5):491-9.
5. Sermet-Gaudelus I, Couderc L, Vrielynck S, et al. Groupe de travail dépistage de la Fédération des centres de ressources et de compétences de la mucoviscidose ; Groupe de travail dépistage de la Fédération des centres de ressources et de compétences de la mucoviscidose. Recommandations nationales pour la prise en charge du nourrisson dépisté atteint de mucoviscidose. Consensus de la Fédération des centres de ressources et de compétences de la mucoviscidose. Arch Pediatr 2014;21(6):654-62.
6. Davies G. Does newborn screening improve early lung function in cystic fibrosis? Paediatr Respir Rev 2022;42:17-22.
7. Martiniano SL, Elbert AA, Farrell PM, et al. Outcomes of infants born during the first 9 years of CF newborn screening in the United States: A retrospective Cystic Fibrosis Foundation Patient Registry cohort study. Pediatr Pulmonol 2021;56(12):3758-67.
8. Seddon L, Dick K, Carr SB, et al. Communicating cystic fibrosis newborn screening results to parents. Eur J Pediatr 2021;180(4):1313-6.
9. Martiniano SL, Wu R, Farrell PM, et al. Late diagnosis in the era of universal newborn screening negatively affects short- and long-term growth and health outcomes in infants with cystic fibrosis. J Pediatr 2023;262:113595.
10. Farrell PM. Why cystic fibrosis newborn screening programs have failed to meet original expectations… thus far. Mol Genet Metab 2023;140(1-2):107679.
11. Munck A, Southern KW, Murphy J, et al. European CF Society Neonatal Screening Working Group (ECFS NSWG). Cystic fibrosis cases missed by newborn bloodspot screening-towards a consistent definition and data acquisition. Int J Neonatal Screen 2023;9(4):65.
12. Audrézet MP, Munck A. Newborn screening for CF in France: An exemplary national experience. Arch Pediatr 2020;27 Suppl 1:eS35-40.
13. Sermet-Gaudelus I, Brouard J, Audrezet MP, et al. Recommandations pour la prise en charge et le suivi des nourrissons pour lesquels un diagnostic de mucoviscidose n’a pu être conclu après dépistage néonatal. Arch Pediatr 2017;24(4):401-14.
14. Hatton A, Bergougnoux A, Zybert K, et al. Reclassifying inconclusive diagnosis after newborn screening for cystic fibrosis. Moving forward. J Cyst Fibros 2022;21(3):448-55.
15. Gonska T, Keenan K, Au J, et al. Outcomes of cystic fibrosis screening-positive infants with inconclusive diagnosis at school age. Pediatrics 2021;148(6):e2021051740.
16. Terlizzi V, Manti S, D’Amico F, et al. Biochemical and genetic tools to predict the progression to Cystic Fibrosis in CRMS/CFSPID subjects: A systematic review. Paediatr Respir Rev 2024;51:46-55.
17. Haute Autorité de santé. Évaluation diagnostique et prise en charge des affections liées ou associées à CFTR. https://urls.fr/UVzgBf
18. Protocole national de diagnostic et de soins (PNDS). Évaluation diagnostique et prise en charge des affections liées ou associées à CFTR. Novembre 2021. https://urls.fr/pwoFwX
19. Barry PJ, Simmonds NJ. Diagnosing cystic fibrosis in adults. Semin Respir Crit Care Med 2023;44(2):242-51.
Dans cet article
- Plus de 2 000 mutations génétiques identifiées
- Dépistage néonatal depuis 2002
- Suivi du nourrisson dépisté : mettre en place prévention et traitements
- Diagnostic non conclu, une prise en charge particulière
- Le diagnostic de mucoviscidose sur symptômes est devenu rare
- Changement de paradigme grâce au dépistage néonatal