Forme rare de neurofibromatose de type 5
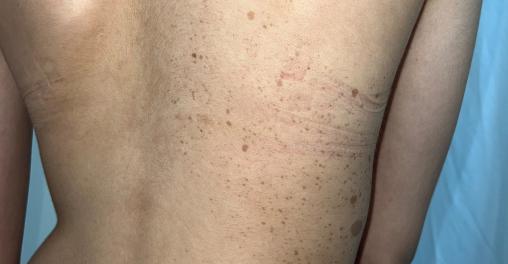
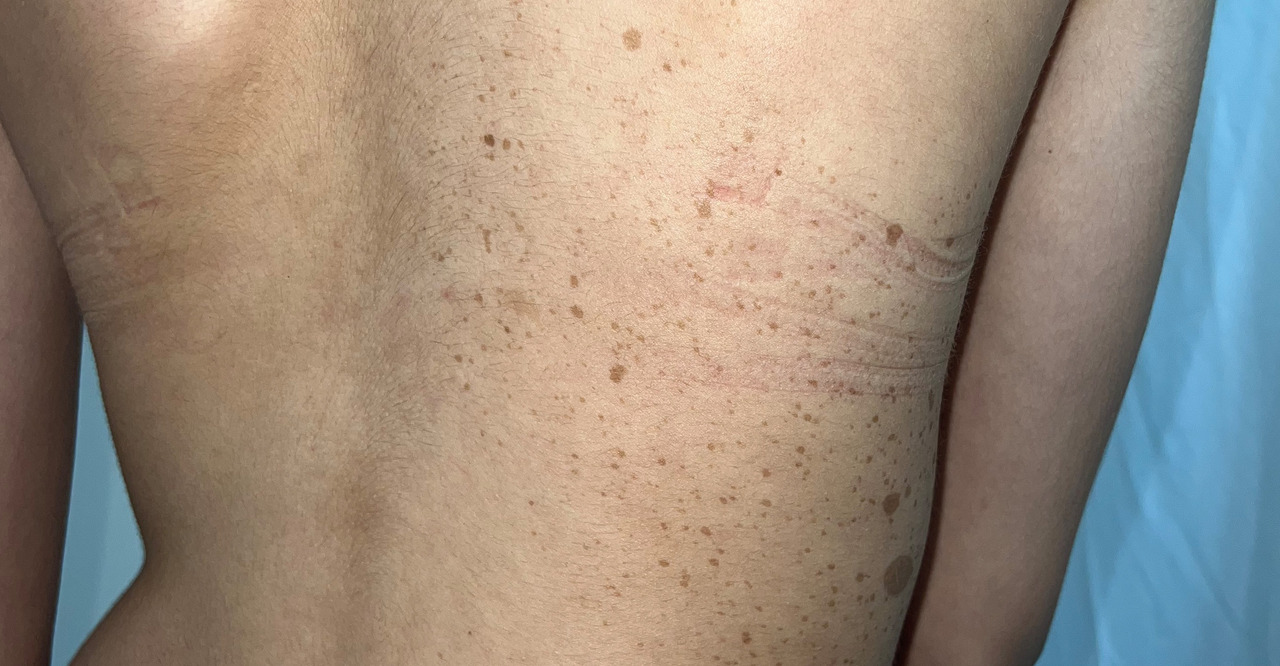
Une patiente de 14 ans, sans antécédents familiaux de neurofibromatose, consulte pour des lésions cutanées évoluant depuis deux ans. Maculeuses, brunâtres, de taille variable, à l’aspect de taches café au lait, elles sont latéralisées en thoraco-abdominal droit, suivant une distribution segmentaire métamérique (figure). L’examen révèle également deux nodules fermes, indolores, de couleur chair, mesurant environ 2 cm de diamètre, localisés au niveau abdominal. Il n’existe pas d’autres signes cutanés ou neurologiques, et aucun signe de dysmorphie n’est observé. L’examen ophtalmologique ne révèle pas de nodules de Lisch.
Une biopsie de l’un des nodules est réalisée, mettant en évidence un neurofibrome circonscrit, non encapsulé et sans signe de malignité.
L’IRM cérébrale et le scanner cervico-thoraco-abdomino-pelvien sont sans anomalie.
Une biopsie de l’un des nodules est réalisée, mettant en évidence un neurofibrome circonscrit, non encapsulé et sans signe de malignité.
L’IRM cérébrale et le scanner cervico-thoraco-abdomino-pelvien sont sans anomalie.
Les neurofibromatoses constituent un groupe hétérogène de maladies affectant principalement le système nerveux et la peau. Il s’agit d’une génodermatose relativement fréquente, caractérisée par la présence de neurofibromes, de taches café au lait ainsi que de certains symptômes neurologiques et ophtalmologiques.
La neurofibromatose de type V, également appelée neurofibromatose segmentaire ou localisée en mosaïque, est une variante rare, caractérisée par des lésions cutanées limitées à un segment circonscrit du corps.1 Le premier cas a été décrit en 1932 par Gammel. Les caractéristiques cliniques classiques de la neurofibromatose segmentaire sont les taches café au lait, les taches de rousseur et/ou les neurofibromes dans une distribution métamérique ou suivant les lignes de Blaschko.2
La maladie peut s’accompagner d’une atteinte systémique et de tumeurs malignes, notamment de tumeurs des gaines des nerfs périphériques et de mélanomes malins, pour les plus fréquentes. D’autres cancers, comme ceux du sein, du côlon, de l’estomac, du poumon et le lymphome de Hodgkin, peuvent aussi être associés. Bien qu’aucune prise en charge spécifique ne soit établie, un diagnostic précis est essentiel pour dépister d’éventuelles complications systémiques.3
Références
1. Dang JD, Cohen PR. Segmental neurofibromatosis and malignancy. Skinmed 2010;8(3):156-9.
2. Victor FC. Segmental neurofibromatosis. Dermatology Online Journal 2005;11(4):20.
3. Chu CH, Chou TC, Liu CI, et al. Multiple segmental neurofibromatosis: Report of two cases with different presentations. Hong Kong J Dermatol Venereol 2016;24:34-9
2. Victor FC. Segmental neurofibromatosis. Dermatology Online Journal 2005;11(4):20.
3. Chu CH, Chou TC, Liu CI, et al. Multiple segmental neurofibromatosis: Report of two cases with different presentations. Hong Kong J Dermatol Venereol 2016;24:34-9
0
« Euh… »
- En savoir plus sur « Euh… »
- Se connecter ou s'inscrire pour poster un commentaire
Syndrome de Parsonage-Turner
Un patient de 43 ans consulte pour une douleur de l’épaule droite (figure) ; il indique que son épaule semble « se décoller ». Dans les antécédents est mentionnée la pratique du volley-ball. À l’examen clinique, les tests de la coiffe de l’épaule se révèlent sans anomalie et sans douleur particulière.
Cependant, lors de l’examen de contre-résistance sur une porte, l’omoplate se décolle du thorax ; on note un coup de vent scapulaire.
Devant ce tableau évocateur d’une dénervation de type syndrome de Parsonage-Turner, un électro-myogramme ainsi qu’une IRM de l’épaule sont réalisés. L’IRM de l’épaule montre une absence de lésion pathologique. L’électromyogramme révèle une atteinte isolée du nerf long thoracique droit avec signes de dénervation abondants. Un traitement par kinésithérapie à type de renforcement musculaire est prescrit. Le patient est réévalué à trois mois avec une récupération partielle.
Cependant, lors de l’examen de contre-résistance sur une porte, l’omoplate se décolle du thorax ; on note un coup de vent scapulaire.
Devant ce tableau évocateur d’une dénervation de type syndrome de Parsonage-Turner, un électro-myogramme ainsi qu’une IRM de l’épaule sont réalisés. L’IRM de l’épaule montre une absence de lésion pathologique. L’électromyogramme révèle une atteinte isolée du nerf long thoracique droit avec signes de dénervation abondants. Un traitement par kinésithérapie à type de renforcement musculaire est prescrit. Le patient est réévalué à trois mois avec une récupération partielle.
Le syndrome de Parsonage-Turner, également appelé névralgie amyotrophique, correspond à une atteinte rare du plexus brachial. Il se manifeste généralement dans un contexte inflammatoire (post-infectieux, post-chirurgical ou post-vaccinal) par une douleur intense et brutale de l’épaule, souvent unilatérale, suivie en quelques jours d’une paralysie puis d’une amyotrophie des muscles scapulaires. L’atteinte motrice et parfois sensitive des troncs nerveux est précisée grâce à l’électromyogramme, pouvant être complétée par une IRM afin d’éliminer des lésions compressives.
Le traitement antalgique à la phase aiguë peut être associé à une corticothérapie. La prise en charge au long cours est fondée sur la rééducation avec renforcement musculaire.
La guérison peut être lente – en plusieurs mois ou plusieurs années (environ trois ans) – et est parfois incomplète. Des récidives sont possibles.
Pour en savoir plus
Conyer RT, Sperling JW. Anatomic total shoulder arthroplasty in a patient with Parsonage-Turner syndrome: a case report. JSES Rev Rep Tech 2023;3(4):540-7.
Meiling JB, Boon AJ, Niu Z, et al. Parsonage-Turner syndrome and Hereditary Brachial Plexus Neuropathy. Mayo Clin Proc 2024;99(1):124-40.
Patterson DL, Deremee RA, Hunt LW. Severe asthma complicated by bilateral diaphragmatic paralysis attributed to Parsonage-Turner syndrome. Mayo Clin Proc 1994;69(8):774-8.
Meiling JB, Boon AJ, Niu Z, et al. Parsonage-Turner syndrome and Hereditary Brachial Plexus Neuropathy. Mayo Clin Proc 2024;99(1):124-40.
Patterson DL, Deremee RA, Hunt LW. Severe asthma complicated by bilateral diaphragmatic paralysis attributed to Parsonage-Turner syndrome. Mayo Clin Proc 1994;69(8):774-8.
0
Maladie de Paget extramammaire
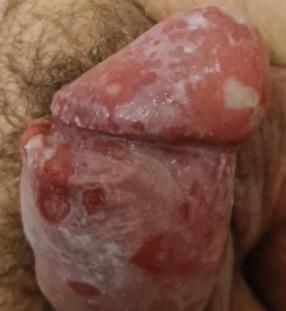
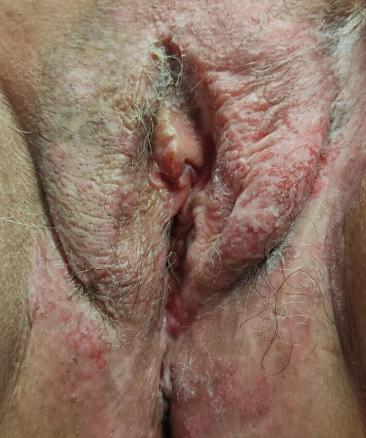
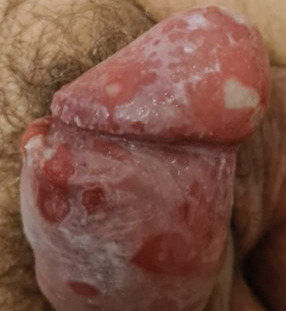
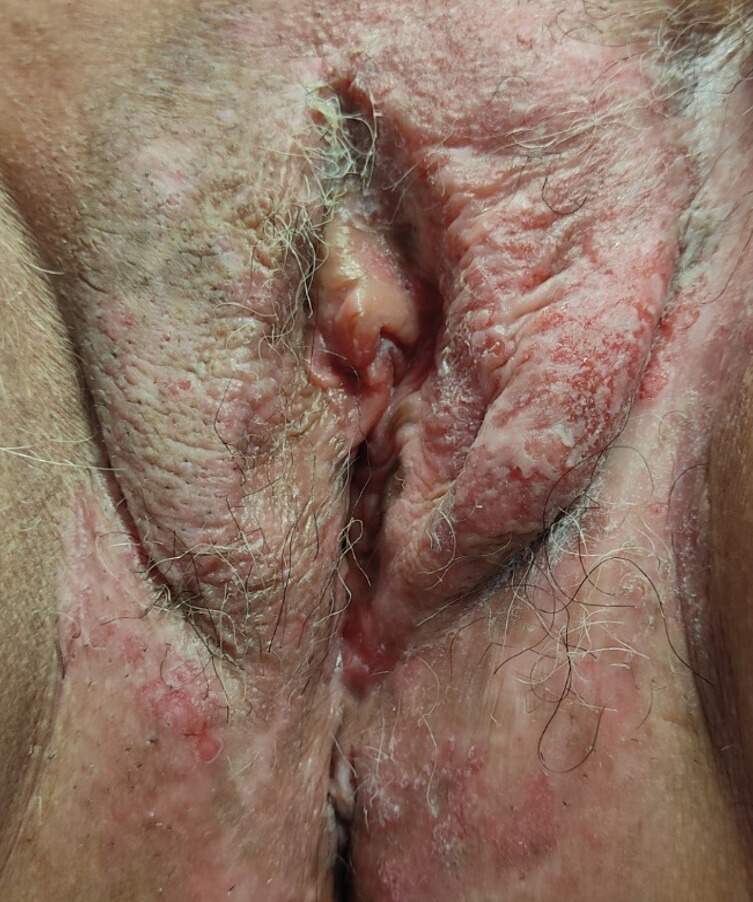
Paul, 70 ans, et Fiona, 54 ans, consultent séparément pour une dermatose génitale mal limitée, fissuraire, douloureuse, d’extension progressive, évoluant depuis plusieurs années et ne répondant à aucun traitement local (ni antimycotiques, ni dermocorticoïdes) [fig. 1 et 2]. Un carcinome colorectal a récemment été diagnostiqué chez l’un d’eux.
La maladie de Paget extramammaire correspond à un adénocarcinome intraépidermique. Deux formes sont à distinguer : les formes primaires, localisées au niveau cutané, mais qui peuvent s’associer dans 2 à 20 % des cas à un adénocarcinome local sous-jacent ; les formes secondaires qui sont satellites d’un adénocarcinome à distance (urothélial ou colorectal le plus souvent).
Cliniquement, il s’agit d’une plaque érythémateuse, plus ou moins érosive et suintante, bien limitée, et d’extension progressive. Des rhagades (ulcérations linéaires) sont présentes en surface. L’aspect clinique peut être trompeur.
Le diagnostic est histologique. Une biopsie cutanée doit être réalisée ainsi qu’un bilan complémentaire (imagerie, endoscopie…), à la recherche d’un adénocarcinome à distance.
La prise en charge repose sur l’exérèse chirurgicale lorsqu’elle est possible. Les récidives locales sont fréquentes (17 à 38 % des cas). D’autres traitements peuvent être proposés, comme la photothérapie dynamique, l’imiquimod ou le laser CO2, mais le risque de récidive est important. La prise en charge de l’adénocarcinome associé, local ou à distance, nécessite le concours d’une équipe spécialisée (gynécologues, urologues, gastroentérologues...).
Pour en savoir plus
Pérez JC, Salgado AC, Pérez-Mies B, et al. Extramammary Paget Disease: A Therapeutic Challenge, for a Rare Entity. Curr Oncol Rep 2023;25(10):1081-94.
0
Lymphome testiculaire primitif
Monsieur X., 72 ans, sans antécédents particuliers, consulte pour une masse testiculaire droite indolore, qui augmente progressivement de volume depuis un mois.
L’examen clinique met en évidence une masse testiculaire droite dure, non douloureuse. Il n’y a pas d’adénopathie périphérique. L’examen neurologique est normal.
Les dosages de l’alphafœtoprotéine (AFP) et de la gonadotrophine chorionique (β-HCG) sont normaux, mais le taux de lactate déshydrogénase (LDH) est 1,5 fois plus élevé que la normale.
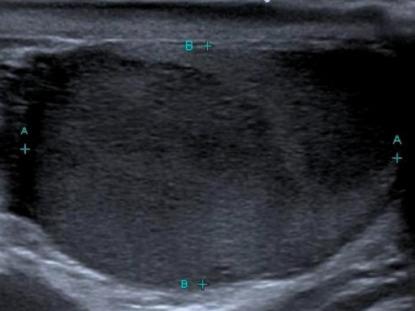
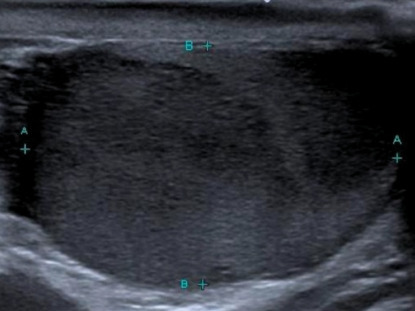
L’échographie révèle une masse hypoéchogène hétérogène du testicule droit (figure). Le TEP-scan montre également une fixation isolée au même niveau, sans atteinte ganglionnaire ni viscérale.
L’IRM cérébrale et la ponction lombaire sont sans anomalies.
Une orchidectomie droite est réalisée. L’analyse histologique révèle un lymphome diffus à grandes cellules B (CD20 +), avec un marqueur de prolifération (Ki67) à 90 %, sans envahissement de l’albuginée ni de l’épididyme.
Le patient a été traité par six cycles de R-CHOP toutes les trois semaines, puis par radiothérapie prophylactique du testicule controlatéral (30 Gy en 15 fractions). Compte tenu de l’absence de facteurs de risque spécifiques, aucune prophylaxie du système nerveux central n’est mise en place.
Le patient a bien toléré le traitement. Une évaluation post-thérapeutique par TEP-scan a confirmé la rémission complète. À vingt-quatre mois, aucun signe de rechute n’a été détecté.
L’examen clinique met en évidence une masse testiculaire droite dure, non douloureuse. Il n’y a pas d’adénopathie périphérique. L’examen neurologique est normal.
Les dosages de l’alphafœtoprotéine (AFP) et de la gonadotrophine chorionique (β-HCG) sont normaux, mais le taux de lactate déshydrogénase (LDH) est 1,5 fois plus élevé que la normale.
L’échographie révèle une masse hypoéchogène hétérogène du testicule droit (figure). Le TEP-scan montre également une fixation isolée au même niveau, sans atteinte ganglionnaire ni viscérale.
L’IRM cérébrale et la ponction lombaire sont sans anomalies.
Une orchidectomie droite est réalisée. L’analyse histologique révèle un lymphome diffus à grandes cellules B (CD20 +), avec un marqueur de prolifération (Ki67) à 90 %, sans envahissement de l’albuginée ni de l’épididyme.
Le patient a été traité par six cycles de R-CHOP toutes les trois semaines, puis par radiothérapie prophylactique du testicule controlatéral (30 Gy en 15 fractions). Compte tenu de l’absence de facteurs de risque spécifiques, aucune prophylaxie du système nerveux central n’est mise en place.
Le patient a bien toléré le traitement. Une évaluation post-thérapeutique par TEP-scan a confirmé la rémission complète. À vingt-quatre mois, aucun signe de rechute n’a été détecté.
Le lymphome testiculaire primitif (LTP) est une entité rare, représentant moins de 2 % des lymphomes non hodgkiniens et environ 5 % des tumeurs testiculaires.1 Il s’agit le plus souvent d’un lymphome diffus à grandes cellules B, touchant principalement les sujets âgés. Il se manifeste par une masse testiculaire, généralement unilatérale, indolore. Il est souvent diagnostiqué tardivement en raison de son aspect trompeur, mimant une tumeur germinale. Il faut y penser devant une masse testiculaire chez un homme âgé de plus de 60 ans, surtout si les marqueurs tumoraux sont normaux.
La prise en charge repose sur l’orchidectomie, suivie d’une chimiothérapie de type R-CHOP, d’une radiothérapie prophylactique du testicule controlatéral, et parfois d’une prophylaxie du système nerveux central (SNC).
Le pronostic dépend du stade initial, de l’âge et de la réponse au traitement. Il reste réservé, comparé à celui des autres lymphomes diffus à grandes cellules B. Le LTP se caractérise par un risque élevé de rechute, parfois tardive, dans des sites extranodaux (testicule controlatéral, SNC, séreuses).1,2 Le suivi doit donc être prolongé, avec une vigilance sur le plan neurologique. Une approche multidisciplinaire est indispensable pour une prise en charge optimale.
Références
1. Dabaja BS, Specht L, Yahalom J. Lymphoblastic Lymphoma: Guidelines from the International Lymphoma Radiation Oncology Group (ILROG). Int J Radiat Oncol Biol Phys 2018;102(3):508-14.
2. Zouhair A, Herrmann E, Ugurluer G, et al. Primary testicular lymphoma. Swiss Med Wkly 2010;140:w13076.
2. Zouhair A, Herrmann E, Ugurluer G, et al. Primary testicular lymphoma. Swiss Med Wkly 2010;140:w13076.
0
Accident vasculaire cérébral ischémique révélant un thrombus intraventriculaire gauche
Un patient âgé de 57 ans, sans antécédent notable, est vu aux urgences pour un syndrome déficitaire de l’hémicorps droit avec mutisme d’installation brutale. L’examen neurologique objective une hémiparésie droite avec aphasie de Broca. Le reste de l’examen clinique, y compris cardiovasculaire, est sans particularité.
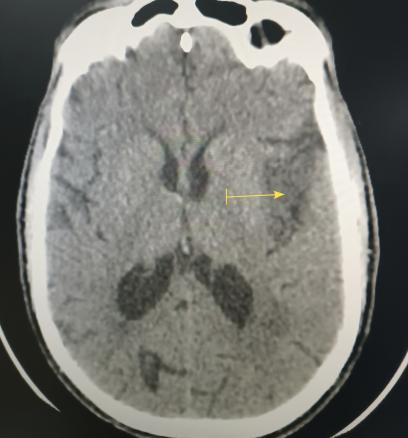
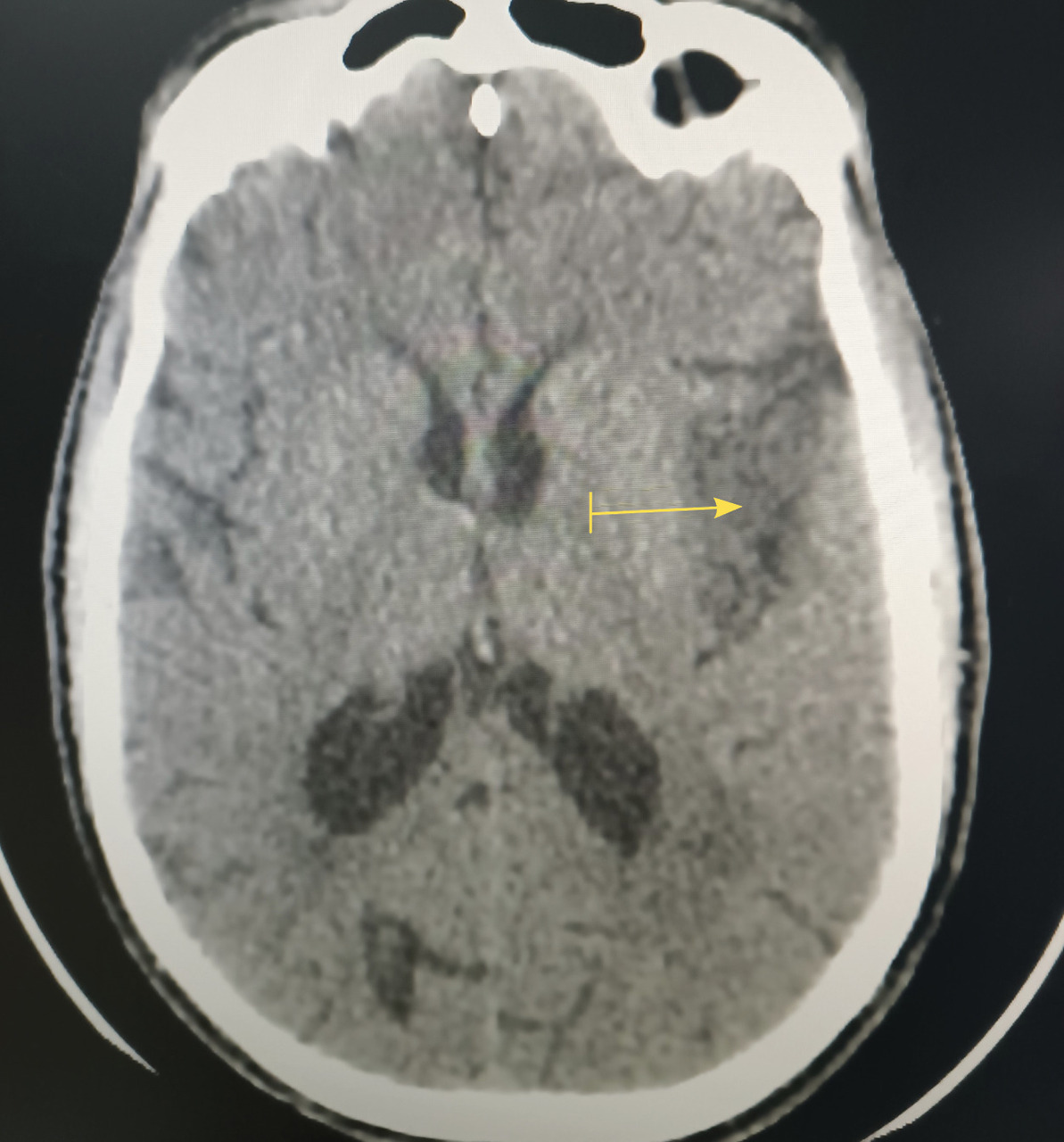
Le scanner cérébral révèle un accident vasculaire cérébral ischémique du territoire de l’artère cérébrale moyenne superficielle gauche (fig. 1).
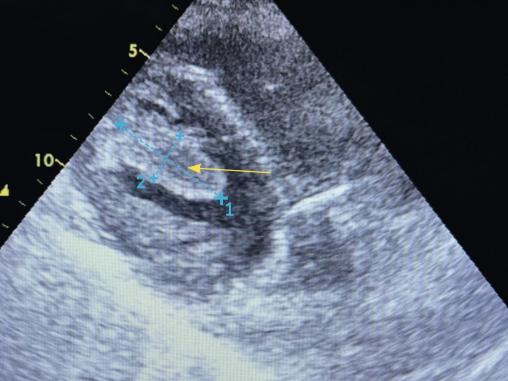
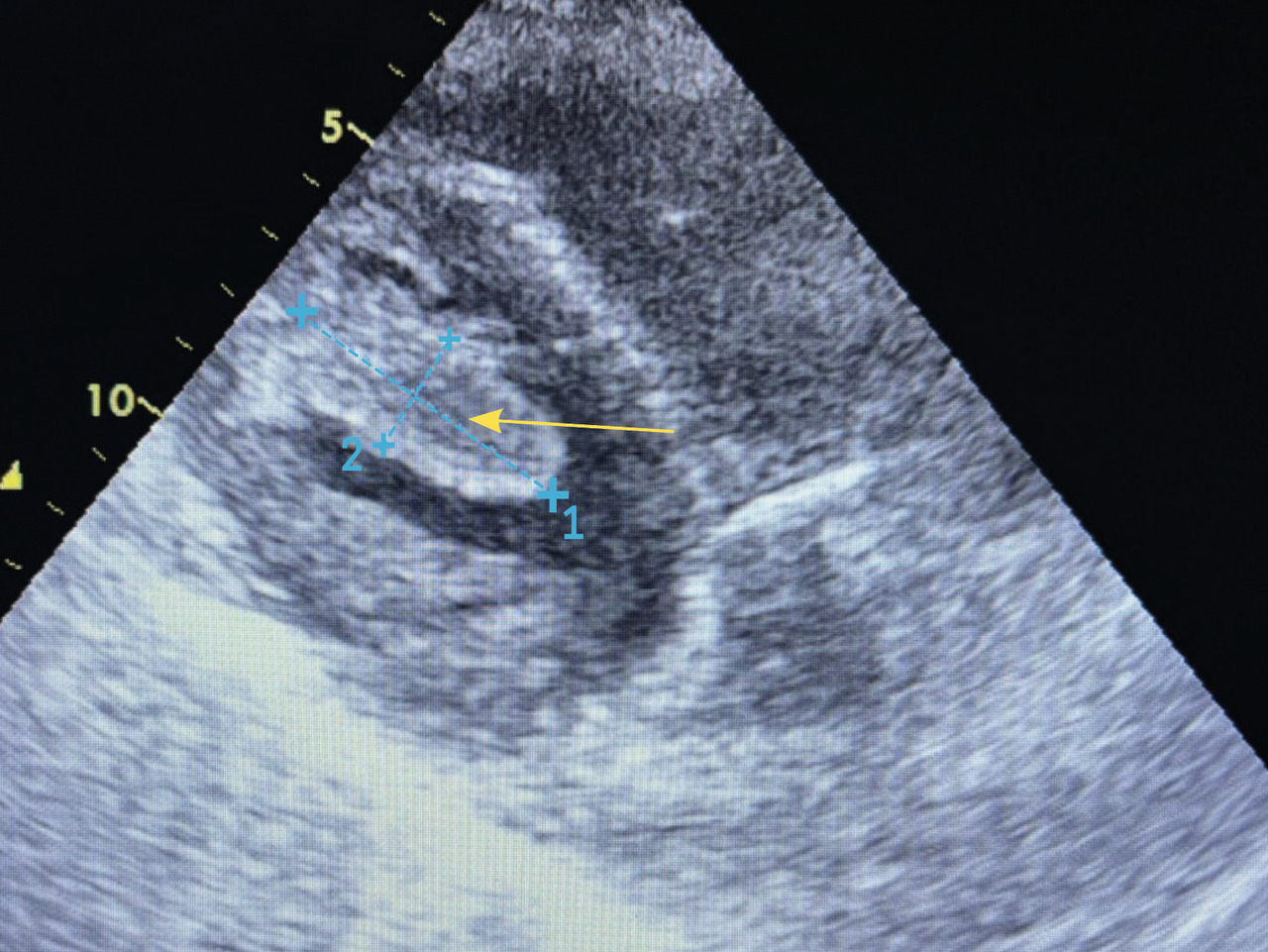
Une échocardiographie transthoracique montre la présence d’un thrombus intraventriculaire gauche (VG) apical (fig. 2), siège d’une akinésie de l’apex et une fonction globale conservée avec fraction d’éjection du VG (FEVG) estimée à 50 %.
Le scanner cérébral révèle un accident vasculaire cérébral ischémique du territoire de l’artère cérébrale moyenne superficielle gauche (fig. 1).
Une échocardiographie transthoracique montre la présence d’un thrombus intraventriculaire gauche (VG) apical (fig. 2), siège d’une akinésie de l’apex et une fonction globale conservée avec fraction d’éjection du VG (FEVG) estimée à 50 %.
Le thrombus intraventriculaire gauche survient principalement chez les patients ayant une dysfonction systolique importante ; sa formation dans un VG normal est rare.1 L’akinésie et la dyskinésie de la paroi du VG entraînent une stase sanguine qui augmente le risque de formation d’un thrombus, surtout au niveau de l’apex. La présence d’un thrombus intraventriculaire augmente le risque de survenue d’une complication thromboembolique artérielle, notamment les accidents vasculaires cérébraux ischémiques et les embolies périphériques.2
Parmi les examens complémentaires, l’échocardiographie transthoracique est considérée comme l’examen clé pour évaluer la fonction cardiaque et détecter le thrombus. Toutefois, une imagerie par résonance magnétique cardiaque avec injection de gadolinium semble avoir une meilleure spécificité et sensibilité pour la détection d’un thrombus.
Références
1. Kawamoto J, Ishibashi K, Shibukawa T, et al. Left ventricular thrombus with a normal heart. Gen Thorac Cardiovasc Surg 2007;55(8):322-4.
2. Lattuca B, Silvain J. Thrombus intraventriculaire : diagnostic et traitement. Real Cardiol 2023;380:28-34.
2. Lattuca B, Silvain J. Thrombus intraventriculaire : diagnostic et traitement. Real Cardiol 2023;380:28-34.
0