Nos petites habitudes, les bonnes… comme les mauvaises !
- En savoir plus sur Nos petites habitudes, les bonnes… comme les mauvaises !
- Se connecter ou s'inscrire pour poster un commentaire
Kératoacanthome
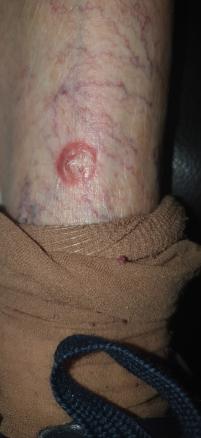
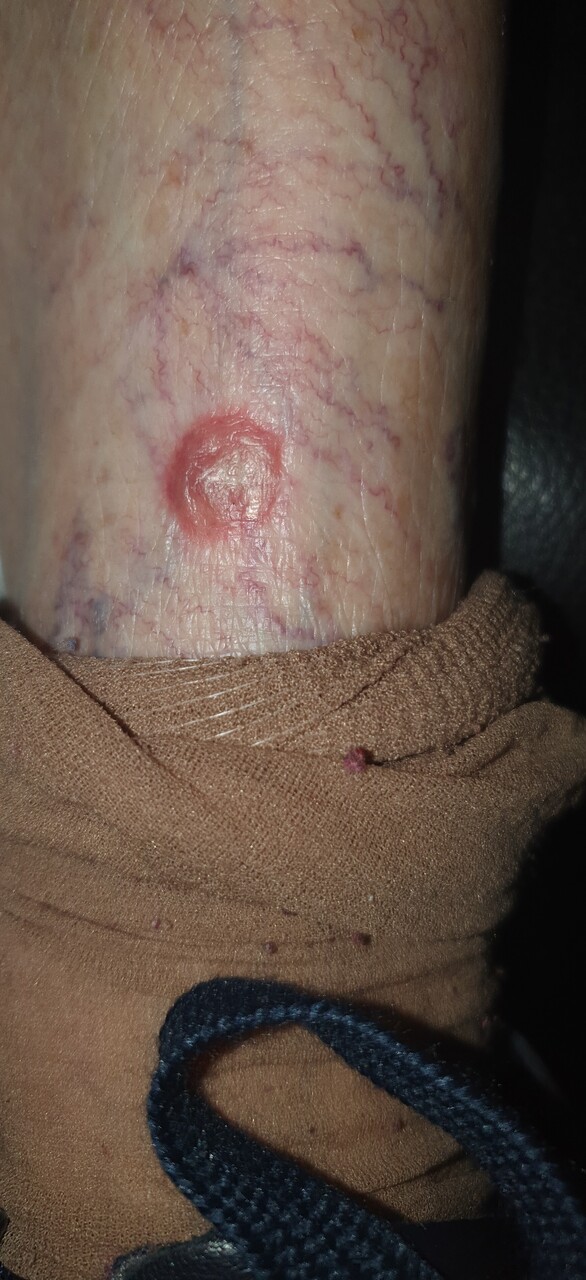
Yvette, 87 ans, consulte pour une formation nodulaire au centre croûteux apparue au niveau de sa jambe gauche (figure), de développement rapide et qui s’étend.
Le kératoacanthome est considéré comme un type de carcinome épidermoïde dont le potentiel de malignité est faible. Il touche les patients de plus de 50 ans à phototype clair. Plusieurs facteurs favorisants sont incriminés : photo-exposition intense, infection à papillomavirus (HPV), exposition à certaines substances chimiques (arsenic…).
Cliniquement, les kératoacanthomes apparaissent au niveau du cou, de la partie supérieure du thorax, des mains, bras et avant-bras, et des jambes. Ils évoluent en trois phases : prolifération, maturité et résorption. Typiquement, la lésion se caractérise par un nodule de couleur chair se développant très rapidement (phase de prolifération, parfois douloureuse). Au centre, un bouchon kératosique est mis en évidence ; il peut s’agir d’une dépression centrale prenant l’aspect d’un volcan. Le diamètre d’un kératoacanthome varie entre 0,5 et 2 cm.
La prise en charge repose, en premier lieu, sur l’analyse anatomopathologique de la tumeur. Même si le diagnostic semble évident, des formes invasives de carcinomes épidermoïdes peuvent prendre le même aspect, raison pour laquelle l’exérèse chirurgicale est indispensable.
Pour en savoir plus
Quereux G. Cancers cutanés : quels risques ? Rev Prat Med Gen 2018;32(1004):499-500.
0
Thrombose veineuse cérébrale
Un homme de 19 ans est hospitalisé pour un épisode de confusion brutale. Il a un déficit de l’attention traité par rispéridone et un antécédent maternel de mutation du facteur V de Leiden. L’interrogatoire ne révèle pas de prise de toxiques. L’examen clinique met en évidence un syndrome cérébelleux isolé avec une franche ataxie cérébelleuse et une dysmétrie. Les analyses biologiques sont sans particularités, notamment en toxicologie. L’imagerie par résonance magnétique (IRM) ne met en évidence aucune anomalie expliquant la symptomatologie. Durant son hospitalisation de plusieurs jours, les symptômes du patient régressent spontanément ; l’hypothèse étiologique d’une intoxication non détectée sur les analyses initiales est alors posée.
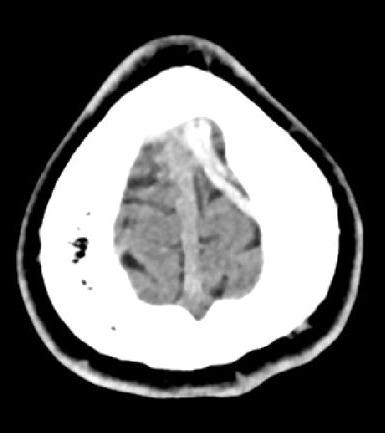
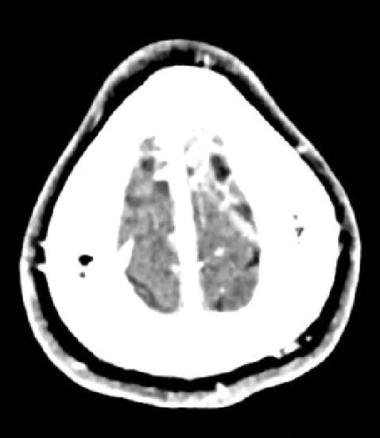
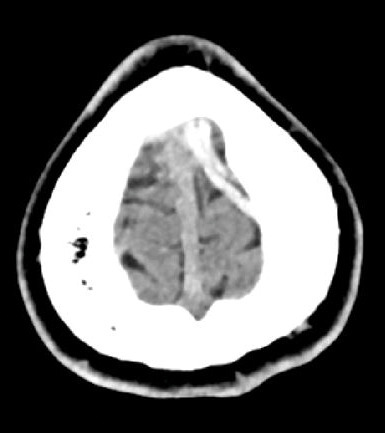
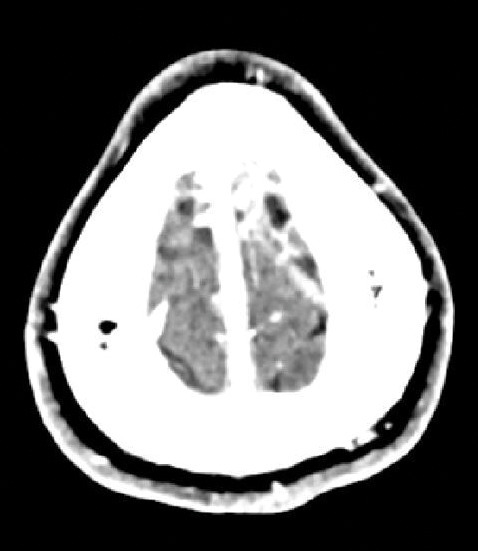
Huit jours plus tard, le patient présente un épisode de clonies du membre supérieur droit, suivi, dans la journée, d’une crise tonico-clonique généralisée, sans céphalées, nausées ni vomissements. Il s’agit du premier épisode décrit chez ce patient. Le scanner cérébral non injecté révèle une hyperdensité au niveau de la veine corticale de la convexité frontale postérieure gauche (fig. 1). L’angioscanner confirme une thrombose veineuse cérébrale (TVC), sans hémorragie ni atteinte parenchymateuse associée (fig. 2). Une relecture de l’IRM confirme l’absence d’antériorité ou de signe qui aurait pu être annonciateur d’une TVC.
Une anticoagulation curative par héparine de bas poids moléculaire et un traitement antiépileptique par lévétiracétam ont été instaurés. Le patient a ensuite été transféré dans une unité de soins intensifs neurovasculaires.
Huit jours plus tard, le patient présente un épisode de clonies du membre supérieur droit, suivi, dans la journée, d’une crise tonico-clonique généralisée, sans céphalées, nausées ni vomissements. Il s’agit du premier épisode décrit chez ce patient. Le scanner cérébral non injecté révèle une hyperdensité au niveau de la veine corticale de la convexité frontale postérieure gauche (fig. 1). L’angioscanner confirme une thrombose veineuse cérébrale (TVC), sans hémorragie ni atteinte parenchymateuse associée (fig. 2). Une relecture de l’IRM confirme l’absence d’antériorité ou de signe qui aurait pu être annonciateur d’une TVC.
Une anticoagulation curative par héparine de bas poids moléculaire et un traitement antiépileptique par lévétiracétam ont été instaurés. Le patient a ensuite été transféré dans une unité de soins intensifs neurovasculaires.
La thrombose veineuse cérébrale (TVC) est une pathologie peu fréquente ; elle représente moins de 1 % des accidents vasculaires cérébraux.1 Elle est souvent diagnostiquée tardivement en raison de sa présentation clinique polymorphe.
Les causes de TVC sont le plus souvent des troubles de l’hémostase, des infections, des traumatismes crâniens, des tumeurs ou des gestes chirurgicaux récents.2
Les localisations préférentielles concernent les sinus latéraux, le sinus longitudinal supérieur ou le sinus caverneux.3 L’atteinte des veines corticales est exceptionnelle.4
L’imagerie permet de poser le diagnostic. Le scanner non injecté peut mettre en évidence des signes directs tels que l’hyperdensité spontanée d’un caillot (signe de la corde). Néanmoins, ce signe n’a pas une spécificité élevé, puisqu’il est aussi observé chez des patients atteints de polyglobulie, chez le sujet jeune avec un hématocrite élevé ou chez le patient déshydraté.5 L’angioscanner met en évidence un défaut de remplissage après injection (signe du delta vide quand le caillot est au niveau sinusal).4 L’IRM constitue l’examen de référence, en particulier pour les formes corticales et les atteintes parenchymateuses.2
Ce cas illustre la difficulté diagnostique des TVC, dont les manifestations initiales peuvent être trompeuses et les anomalies scannographiques discrètes. Il souligne l’importance d’une lecture attentive des coupes apicales au scanner afin d’éviter un retard diagnostique, aux conséquences potentiellement graves.
Références
1. Reiner P, Crassard I, Lukaszewicz AC. Thrombose veineuse cérébrale. Réanimation 2013;22(6):624‑33.
2. Arquizan C. Thrombophlébites cérébrales : aspects cliniques, diagnostic et traitement. Réanimation 2001;10(4):383‑91.
3. Kazouini I, Dahman H, Bouktib Y, et al. Aspects radiologiques de la thrombose veineuse cérébrale : à propos d’une série de 100 cas. J Neuroradiol 2025;52(2):101245.
4. Alami B, Boujraf S, Quenum L, et al. La thrombose veineuse cérébrale : aspects clinico-radiologiques, à propos d’une série de 62 cas. J Med Vasc 2019;44(6):387‑99.
5. Bonneville F. Imagerie des thromboses veineuses cérébrales. J Radiol Diagn Interv 2014;95(12):1130‑5.
2. Arquizan C. Thrombophlébites cérébrales : aspects cliniques, diagnostic et traitement. Réanimation 2001;10(4):383‑91.
3. Kazouini I, Dahman H, Bouktib Y, et al. Aspects radiologiques de la thrombose veineuse cérébrale : à propos d’une série de 100 cas. J Neuroradiol 2025;52(2):101245.
4. Alami B, Boujraf S, Quenum L, et al. La thrombose veineuse cérébrale : aspects clinico-radiologiques, à propos d’une série de 62 cas. J Med Vasc 2019;44(6):387‑99.
5. Bonneville F. Imagerie des thromboses veineuses cérébrales. J Radiol Diagn Interv 2014;95(12):1130‑5.
0
Fracture du triquétrum
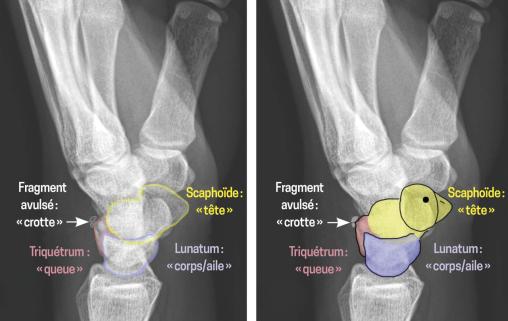
Un homme de 45 ans consulte après une chute accidentelle avec réception sur la main gauche en hyperextension. Il se plaint d’une douleur aiguë au niveau du poignet gauche ; un gonflement est apparu principalement sur le bord ulnaire. L’examen clinique révèle une douleur à la palpation du triquétrum, une limitation fonctionnelle du poignet mais l’absence de déformation visible. Les radiographies standard montrent une petite zone corticale osseuse avulsée sur la projection dorso-latérale du triquetrum.
Une tomodensitométrie confirme une fracture corticale dorsale avec un fragment osseux avulsé.
Une tomodensitométrie confirme une fracture corticale dorsale avec un fragment osseux avulsé.
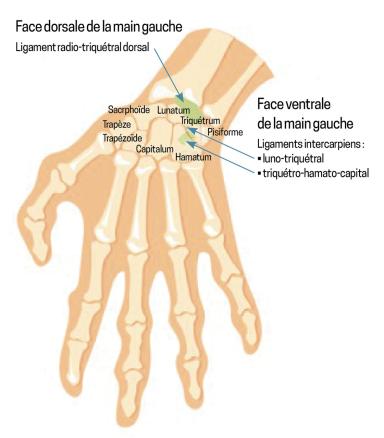
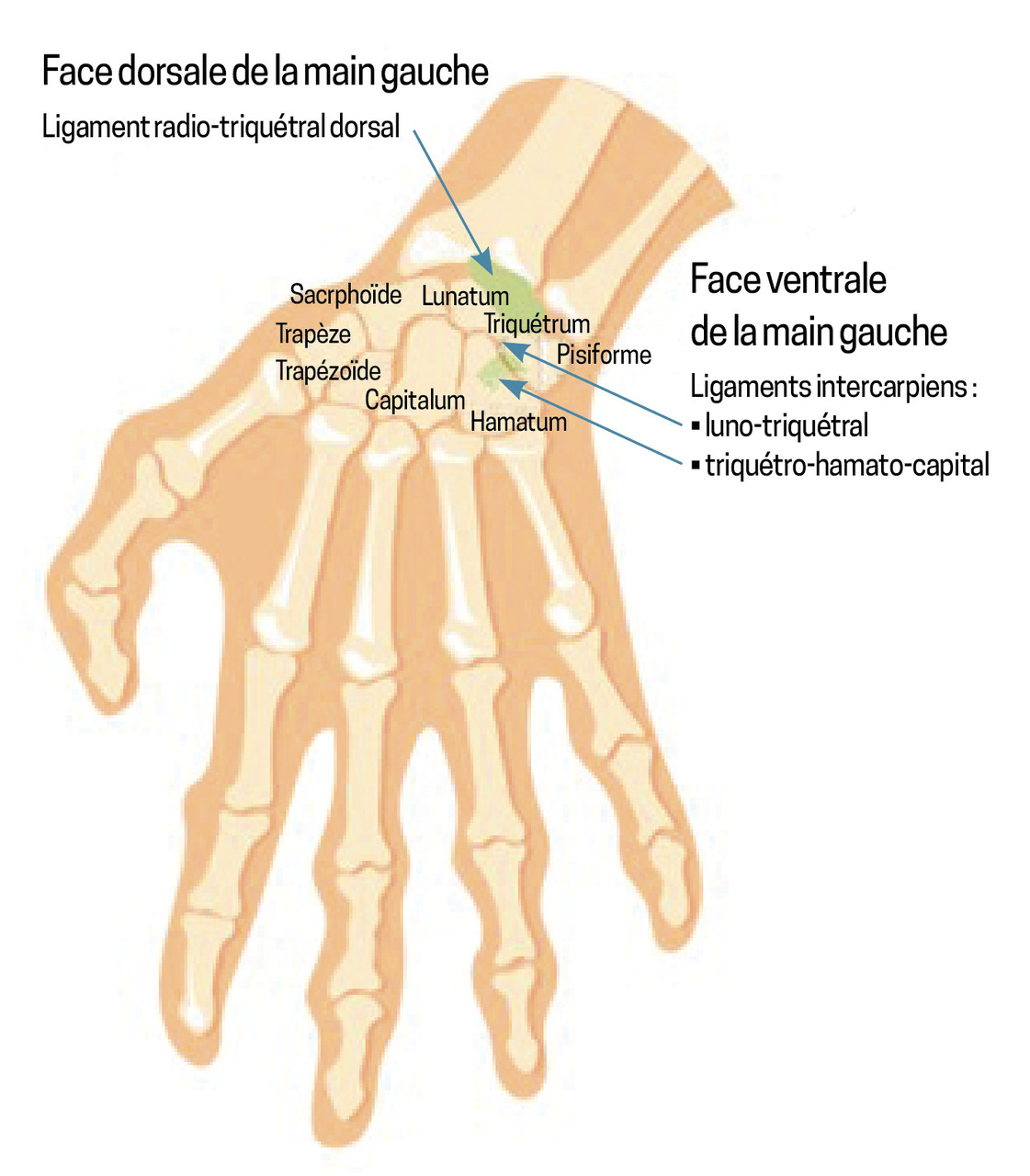
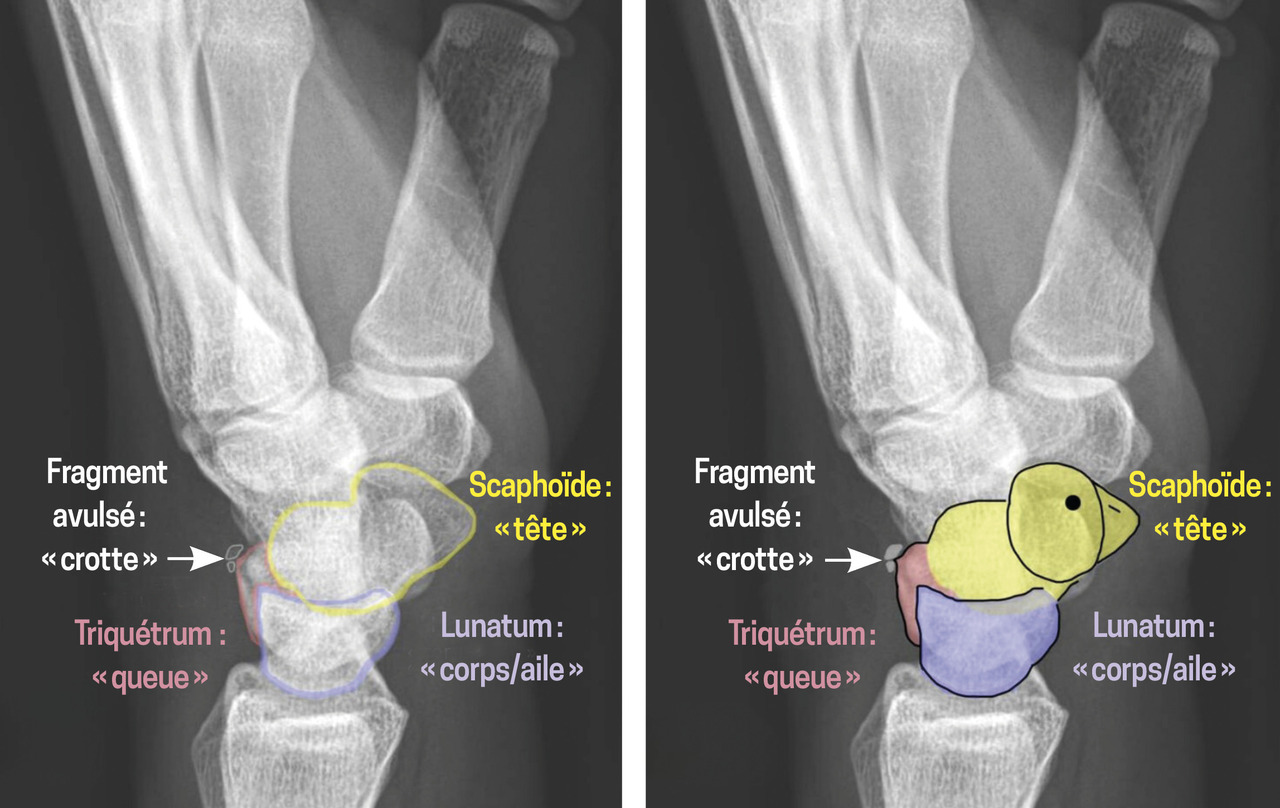
Les fractures du triquétrum sont les deuxièmes fractures carpiennes les plus fréquentes après celles du scaphoïde.1 Elles résultent d’un mécanisme d’impaction ou d’avulsion au niveau des sites d’insertion des ligaments intercarpiens (le triquétro-hamato-capital et le luno-triquétral), ainsi que du ligament radio-triquétral dorsal (fig. 1). Bien que généralement bénignes, ces fractures peuvent entraîner des séquelles fonctionnelles si elles ne sont pas identifiées et traitées. Or, la radiographie de face met rarement en évidence des signes clairs de fracture. En revanche, le « signe du canard qui fait caca » (pooping duck sign) est un repère radiographique spécifique de fracture dorsale du triquétrum en vue latérale : le fragment avulsé représente la « crotte », tandis que le scaphoïde, le lunatum et le triquétrum dessinent respectivement la tête, le corps et la queue du « canard »2 (fig. 2).
Les diagnostics différentiels incluent les fractures du scaphoïde, les luxations périlunaires et les lésions ligamentaires intercarpiennes. Une évaluation clinique rigoureuse et des examens d’imagerie avancés, comme la tomodensitométrie ou l’IRM, sont essentiels pour un diagnostic précis et une prise en charge adaptée.
Le traitement est conservateur, avec une immobilisation plâtrée de six semaines. Une chirurgie d’exérèse peut être envisagée en cas de pseudarthrose douloureuse ou de fragment symptomatique.
Références
1. Guo RC, Cardenas JM, Wu CH. Triquetral Fractures Overview. Curr Rev Musculoskelet Med 2021;14(2):101‑6.
2. Dharmshaktu GS, Dharmshaktu IS, Agarwal N. “Pooping duck” sign. Annals of Medical Science & Research 2023;2(1):56-7.
2. Dharmshaktu GS, Dharmshaktu IS, Agarwal N. “Pooping duck” sign. Annals of Medical Science & Research 2023;2(1):56-7.
0