Mme B., âgée de 71 ans, consulte aux urgences ophtalmologiques pour une sensation de flou visuel en vision de loin bilatéral évoluant depuis quelques semaines, accompagné de douleurs orbitaires et de céphalées peu intenses. Elle a pour antécédents notables une hypertension artérielle traitée par irbésartan et une dyslipidémie traitée par simvastatine. Elle est d’origine grecque et vit en France avec son mari depuis vingt-cinq ans, entourée de ses enfants et petits-enfants. Elle est encore très active et parfaitement autonome.
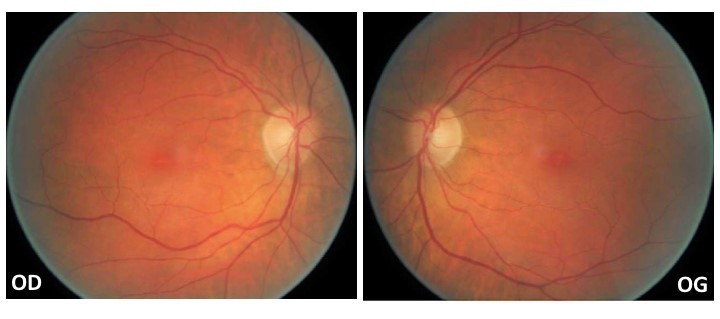
La meilleure acuité visuelle corrigée est préservée à 10/10 Parinaud 2 aux deux yeux. La pression intraoculaire est symétrique à 19 mmHg. L’examen du segment antérieur met en évidence une kératite superficielle bilatérale secondaire à une sécheresse oculaire et l’examen du fond d’œil est le suivant(figure 1) :
Question 1. À l’aide de l’ensemble de ces informations, vous concluez que la gêne visuelle de Mme B. est probablement liée à :
L’aspect du fond d’œil est normal. Voici un exemple de macula rouge cerise chez un patient victime d’occlusion de l’artère centrale de la rétine (figure 2). Le tableau de flou visuel depuis plusieurs mois chez une patiente de 71 ans est très atypique pour une névrite optique. La pression intraoculaire est normale et le nerf optique n’est pas excavé de façon pathologique. Une presbytie donnera une baisse visuelle de près et non pas un flou visuel de loin.
Vous diagnostiquez une kératite bilatérale et prescrivez des soins locaux. Mme B. revient aux urgences quinze jours plus tard car la gêne visuelle continue de s’aggraver malgré les soins de la kératite. Elle se plaint également d’un trouble de la vision des couleurs et de flashs lumineux colorés. La meilleure acuité visuelle mesurée est de 6/10 Parinaud 3 à droite et 7/10 Parinaud 2 à gauche. Votre examen du segment antérieur et du fond d’œil sont normaux. Il n’y a pas de déficit pupillaire afférent relatif.
Question 2. La baisse d’acuité visuelle de Mme B pourrait être liée à :
L’examen ophtalmologique ne serait pas normal si la patiente avait une uvéite postérieure (hyalite) ou une cataracte évoluée. Les raisons d’aggravation rapide des cataractes séniles sont la corticothérapie ou des variations glycémiques rapides chez les diabétiques, ce qui n’est pas son cas. Les migraines avec aura se manifestent par des phosphènes intermittents et non pas permanents, et ne donnent pas de baisse d’acuité visuelle. Il peut s’agir d’une neuropathie optique, par exemple compressive sur une tumeur chiasmatique, ou d’une maculopathie à fond d’œil normal.
Question 3.Quel(s) examen(s) complémentaire(s) demandez-vous en première intention pour orienter votre diagnostic ?
Le champ visuel est indispensable pour préciser la topographie de l’atteinte (rétine, nerf optique, chiasma, rétro-chiasmatique) et l’OCT est indispensable à la recherche d’une maculopathie à fond d’œil normal. La vision des couleurs permet d’argumenter le diagnostic de neuropathie optique (axe rouge/vert) ou de maculopathie (axe bleu/jaune) et de document la plainte de Mme B. Le test de Lancaster et la CRP n’ont aucune utilité ici.
L’OCT est normale, sans anomalie au niveau de la macula ni du nerf optique.
Question 4. À ce stade, que vous pouvez-vous conclure ?
Dans toute neuropathie optique récente l’OCT sera normale. Elle peut parfois montrer un œdème papillaire dans certaines neuropathies optiques (ischémiques…). La perte en fibres sur l’OCT ne sera visible que trois mois environ après l’épisode de névrite optique ou le début de la neuropathie optique. Cependant une OCT normale ne permet pas d’éliminer une neuropathie optique récente ou peu sévère. Il existe des maculopathies (ou rétinopathies) à OCT normale et il est dans ce cas nécessaire de compléter l’exploration par un ERG.
Question 5. Que signe l’absence de déficit pupillaire afférent relatif ?
La présence d’un déficit pupillaire afférent relatif signe une asymétrie d’information lumineuse transmise par un nerf optique RELATIVEMENT à l’autre. Sa présence signe une neuropathie optique unilatérale ou asymétrique. En effet, en cas de neuropathie optique bilatérale et symétrique, la même quantité réduite d’information lumineuse sera transmise par les deux nerfs optiques et la réaction pupillaire sera donc identique des deux côtés. Il n’y a pas de déficit pupillaire afférent relatif (DPAR) dans les maculopathies ni d’atteintes chiasmatiques ou rétro-chiasmatiques (sauf de rares exceptions).
Rassurés par la normalité de l’OCT vous programmez un champ visuel quinze jours plus tard. Lorsque Mme B. revient pour réaliser l’examen, son état s’est encore dégradé. Elle est accompagnée par sa fille qui vous signale une dégradation de son état général avec une majoration de sa baisse visuelle mais également l’apparition de grandes difficultés à la marche et d’une perte d’autonomie. Elle ne comprend plus le grec qui est pourtant sa langue maternelle. Son acuité visuelle est limitée à une perception lumineuse à droite et inférieure à 1/10 à gauche. Elle est incapable de lire et n’arrive pas à réaliser le champ visuel. L’examen ophtalmologique ainsi que les réflexes photomoteurs sont toujours normaux.
Question 6. Quel examen complémentaire demandez-vous devant l’évolution des symptômes ?
L’aggravation de la baisse d’acuité visuelle majeure avec un réflexe photomoteur préservé, accompagnée d’une alexie, de troubles de la marche et de troubles du langage oriente vers une atteinte encéphalique corticale responsable d’un tableau de cécité corticale progressive. L’IRM cérébrale est donc l’examen clé.
L’IRM cérébrale en séquences T1, T2, FLAIR, et T1 avec injection de gadolinium retrouve une atrophie cortico-sous-corticale marquée du cortex occipital. Vous hospitalisez Mme B. en neurologie. Votre examen clinique retrouve un tableau évocateur de cécité corticale accompagnée d’un syndrome cérébelleux et de myoclonies.
Question 7. Quels signes de votre examen clinique vous ont orienté vers une atteinte du cortex occipital et que recherchez-vous d’autre pour localiser l’atteinte au cortex occipital ?
Une atteinte du cortex occipital peut se manifester par un tableau de cécité corticale qui est défini par une baisse visuelle majeure avec un réflexe photomoteur préservé. D’autres signes peuvent être observés parmi lesquels le plus important à rechercher est la simultagnosie qui traduit l’impossibilité à analyser une scène dans son ensemble, le patient étant focalisé sur les détails. Cette simultagnosie peut être associée à une ataxie optique (errance du regard) et à une apraxie oculaire (difficulté à réaliser des gestes visuo-guidés) qui constituent la triade du syndrome de Balint. La prosopagnosie, qui traduit l’altération des capacités de reconnaissance des visages, correspond à une atteinte du cortex temporal.
Question 8. Quels diagnostics évoquez-vous devant ce tableau de démence rapidement évolutive associant une atteinte du cortex occipital et du cervelet ?
Certaines formes de maladie d’Alzheimer débutent par une atteinte du cortex occipital et le tableau clinique est alors dominé par des troubles neurovisuels et non les classiques troubles mnésiques, qui surviendront plus tardivement. Dans tous les cas, la maladie d’Alzheimer est une pathologie dégénérative d’évolution lente. Ce diagnostic ne peut donc pas être évoqué chez Mme B. en raison de la rapidité d’évolution des troubles. Les diagnostics à évoquer devant des troubles neurologiques centraux subaigus sont les encéphalites dont les causes peuvent être variées (auto-immunes, inflammatoires [mais l’IRM est franchement anormale dans ce cas], paranéoplasiques, infectieuses ou à prions).
Question 10. Quel est l’examen essentiel au diagnostic d’encéphalite à prions ou maladie de Creutzfeldt-Jakob ?
Il n’existe pas d’anticorps anti-prions.
Question 11. Quels sont les éléments cliniques et paracliniques qui vous orientent (ou orienteraient) vers le diagnostic de maladie de Creutzfeldt-Jakob ?
La maladie de Creutzfeldt-Jakob se révèle le plus souvent par un syndrome cérébelleux subaigu parfois accompagné de myoclonies. Certaines formes plus rares se révèlent par des troubles visuels. Il s’agit d’une des hypothèses diagnostiques à évoquer devant un syndrome cérébelleux, des mouvements anormaux ou un tableau de démence d’évolution rapide. Les diagnostics différentiels principaux sont les autres causes d’encéphalites (toxiques, métaboliques, paranéoplasiques, ou infectieuses autres). Il s’agit d’une maladie le plus souvent sporadique. Le bilan paraclinique révèle des ondes lentes périodiques typiques sur l’EEG, un hypersignal en séquence FLAIR/T2 et/ou diffusion du cortex cérébral et des ganglions de la base et la présence de la protéine 14-3-3 dans le LCR. Les pointes centro-temporales lentes diphasiques sont retrouvées dans l’épilepsie à paroxysme rolandique de l’enfant.
Question 12. Le résultat positif de la protéine 14.3.3, l’aspect de l’EEG et de la nouvelle IRM cérébrale sont en faveur de votre suspicion diagnostique de maladie de Creutzfeldt-Jakob. Quelles mesures mettez-vous immédiatement en place ?
La maladie de Creutzfeldt-Jakob est une maladie à déclaration obligatoire devant faire l’objet d’une fiche de déclaration obligatoire anonymisée et d’une fiche de signalement nominative, afin que des mesures immédiates soient mises en place pour éviter sa transmission. Il est indispensable de rechercher des antécédents de procédures invasives (endoscopiques) ou d’interventions chirurgicales dans les six mois précédant le diagnostic afin de prévenir les centres médicaux impliqués et que le matériel soit détruit. Il n’y a pas de transmission directe via le contact humain, un isolement de contact n’est donc pas nécessaire.
Question 13. Inquiète, la famille vous demande de lui expliquer les modalités de transmission : ?
L’évaluation du risque de transmission à l’homme est difficile car la nature précise de l’agent infectieux n’est pas identifiée avec certitude. La plupart (80 %) des cas de Creutzfeldt-Jakob sont des formes sporadiques et les formes génétiques ou acquises (greffe de cornée, maladie de la vache folle, injection d’hormone de croissance, kuru) sont rares. Tous les tissus humains n’ont pas le même risque de transmission et les tissus les plus à risque sont le cerveau, la moelle épinière, la dure-mère et l’œil (rétine et nerf optique surtout). Les transmissions sanguines sont rares. L’hormone de croissance humaine présente également un risque de transmission et est donc interdite à la commercialisation. Il n’y a pas de risque de transmission via la salive ni le contact humain direct.
Question 14. Quelle prise en charge pouvez-vous proposez à Mme B. ?
Il n’existe pas de traitement spécifique de la maladie de Creutzfeldt-Jakob et la prise en charge repose principalement sur l’organisation de soins palliatifs et des mesures d’accompagnement social et psychologique du patient et de sa famille.
Question 15. La famille vous demande quel en est le pronostic ?
De forme acquise ou sporadique, la maladie de Creutzfeldt-Jakob évolue de manière inévitable vers le décès du patient.



Voici un exemple de macula rouge cerise chez un patient victime d’occlusion de l’artère centrale de la rétine (figure 2).
Le tableau de flou visuel depuis plusieurs mois chez une patiente de 71 ans est très atypique pour une névrite optique.
La pression intraoculaire est normale et le nerf optique n’est pas excavé de façon pathologique.
Une presbytie donnera une baisse visuelle de près et non pas un flou visuel de loin.