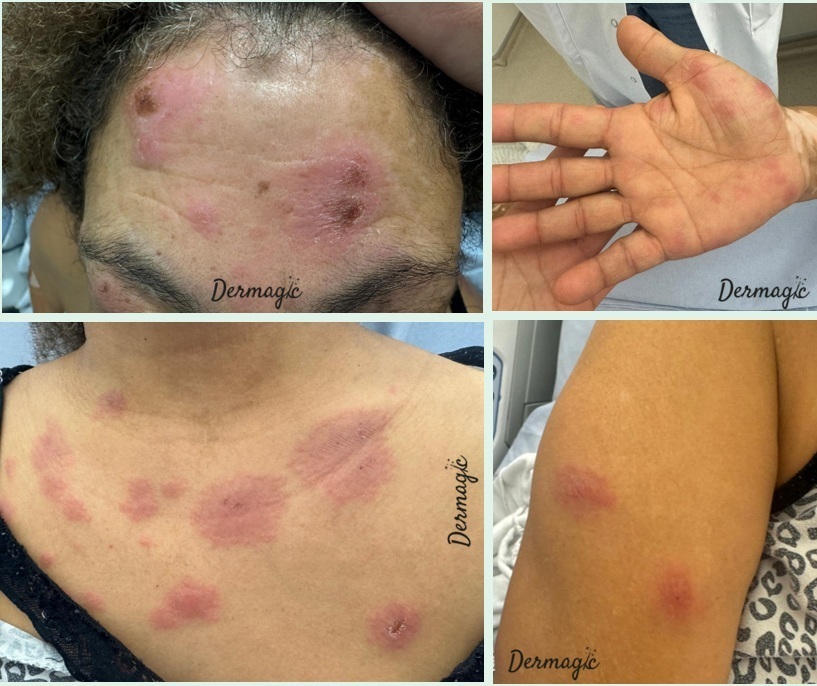
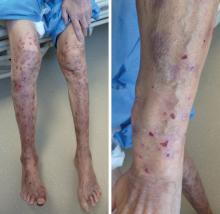
Vous recevez en consultation une femme de 50 ans. Elle est suivie en dermatologie pour un vitiligo. Quinze jours auparavant, elle a eu un syndrome pseudogrippal avec prise de phytothérapie cyprès-échinacée. Cinq jours après, elle a noté l’apparition de cette éruption érythémateuse et œdémateuse des 4 membres, palmoplantaire, faciale et thoracique avec hyperthermie et altération de l’état général puis conjonctivite bilatérale. L’examen montre une hyperlymphocytose à PNN (7,5 G/L) et une CRP à 200 mg/L.
Quel est votre diagnostic ?
L’érythème polymorphe est une dermatose aiguë, souvent post-infectieuse, caractérisée par des lésions en cocarde typiques (cibles à trois zones), symétriques, siégeant préférentiellement sur les extrémités (dos des mains, pieds, avant-bras). Il touche surtout l’adulte jeune. La cause principale est l’infection à HSV (herpès simplex), suivie des infections à Mycoplasma pneumoniae chez l’enfant. Il existe des formes mineures (peu ou pas d’atteinte muqueuse) et des formes majeures (atteinte muqueuse marquée, parfois sévère). Les formes médicamenteuses sont rares mais doivent faire rechercher des toxidermies graves. Le diagnostic est clinique, la biopsie n’est utile qu’en cas de doute. Le traitement repose sur la prise en charge de la cause (traitement antiviral si herpès récurrent), des soins locaux et parfois une corticothérapie courte en cas de formes étendues ou invalidantes.
Il s’agit ici effectivement d’un syndrome de Sweet, ou dermatose aiguë fébrile neutrophilique (dermatose neutrophilique la plus fréquente). Il se manifeste par une apparition brutale de papulonodules érythématoviolacés, douloureux, souvent mamelonnés, parfois bulleux ou pustuleux (chez l'enfant), siégeant préférentiellement sur le visage, le cou et les membres supérieurs. Un syndrome pseudogrippal ou une infection ORL précède fréquemment l’éruption. On distingue plusieurs formes cliniques : la forme classique idiopathique (deux tiers des cas, femmes autour de 40 ans, pic au printemps et à l’automne), les formes para-inflammatoires (liées à des MICI, maladies auto-immunes ou infectieuses), les formes néoplasiques (20 %, surtout hémopathies myéloïdes) et les formes médicamenteuses (G-CSF +++, azathioprine, rétinoïdes). Une atteinte extracutanée (50 % en cas d’hémopathie) peut concerner divers organes. Le diagnostic repose sur la clinique, une NFS (souvent hyperleucocytose à PNN), une biopsie cutanée et un bilan d’extension (TDM TAP, ± myélogramme ou bilan digestif). Le traitement de référence repose sur une corticothérapie systémique (prednisone 0,5 à 1 mg/kg/j), avec décroissance progressive sur 4 à 6 semaines.
Le syndrome de Stevens-Johnson (SJS) est une toxidermie grave, engageant le pronostic vital, caractérisée par une nécrolyse épidermique partielle (atteinte < 10 % de la surface corporelle), associée à une atteinte muqueuse sévère [buccale, oculaire, génitale]). Il survient typiquement 1 à 3 semaines après l’introduction d’un médicament, les plus fréquents étant les antiépileptiques (lamotrigine, carbamazépine), les antibiotiques (sulfamides, β-lactamines) et les AINS. Le début est brutal, avec fièvre, altération de l’état général, puis éruption maculopapuleuse érythémateuse, rapidement bulleuse et douloureuse. L’évolution peut aller vers un décollement cutané, des complications infectieuses, respiratoires ou hématologiques. Le diagnostic est clinique, confirmé par une biopsie cutanée (apoptose kératinocytaire, clivage sous-épidermique). La prise en charge repose sur l’arrêt immédiat du médicament suspect, une hospitalisation en milieu spécialisé (réanimation ou unité de brûlés), des soins locaux intensifs, et parfois des traitements immunomodulateurs (corticoïdes, ciclosporine, IVIG).
L’urticaire aiguë hémorragique est une vascularite leucocytoclasique bénigne, survenant principalement chez le nourrisson et le jeune enfant (6 mois à 4 ans). Elle se manifeste brutalement par des lésions purpuriques annulaires ou arciformes, œdémateuses, parfois spectaculaires, associées à des œdèmes déclives (paupières, mains, pieds) dans un contexte fébrile modéré. Le tableau peut faire craindre une méningococcémie, mais l’état général de l’enfant reste globalement bon. Une infection virale récente ou une vaccination est souvent retrouvée dans les antécédents. Le diagnostic est clinique, soutenu par une biopsie en cas de doute (vascularite des petits vaisseaux avec extravasation d’hématies, sans atteinte viscérale). L’évolution est spontanément favorable en 1 à 3 semaines, sans traitement spécifique, sauf soins symptomatiques (antalgiques, antihistaminiques si prurit).
Les vascularites cutanées correspondent à une inflammation des vaisseaux dermiques, le plus souvent des petits vaisseaux (capillaires, veinules post-capillaires). Elles se manifestent typiquement par un purpura palpable non déclive d’apparition brutale, parfois associé à des bulles, des ulcérations ou des nodules. Le purpura vasculaire est volontiers symétrique, localisé aux membres inférieurs. Les causes sont variées : infections, médicaments, connectivites, cancers ou vascularites systémiques (ex. : purpura rhumatoïde, vascularite urticarienne, cryoglobulinémie). Une biopsie cutanée profonde est indispensable pour confirmer le diagnostic (vascularite leucocytoclasique des petits vaisseaux) et une biologie dirigée permet d’orienter l’étiologie (NFS, VS/CRP, bilan rénal, immunologie, sérologies, cryoglobulinémie). Le traitement dépend de la cause : arrêt du médicament en cas de toxidermie, antibiothérapie si infection, ou traitement immunosuppresseur dans les formes systémiques sévères.
Par le Dr Maud Pellant, interne en dermatologie au CH de Quimper, le Dr Anne-Laure Messagier, PH au CH de Quimper, et le Dr Claire Jacquin-Porretaz, dermatologue libérale.
Plus d’infos sur https://dermagic.fr, le site d’aide à la pratique de la dermatologie réservé aux professionnels de santé.



Plus d’infos sur https://dermagic.fr, le site d’aide à la pratique de la dermatologie réservé aux professionnels de santé.